The revised Common Rule requires consent information to “begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research” (45 CFR 46.116(a)(5)(i)). FDA’s proposed regulations would add identical language to 21 CFR 50.20. Draft guidance from the Food and Drug Administration (FDA) and Office of Human Research Protections (OHRP), issued in 2024, outlined the elements of key information that should precede the presentation of the full informed consent document.
The UI IRB Consent Summary of Key Information template in HawkIRB contains specific instructions and template language to help UI researchers comply with this regulatory guidance.
This educational tool is provided to offer guidance about different approaches to the consent process, including ways to present key information to potential subjects, and document formatting examples.
Facilitating comprehension in the consent process might include:
- Utilizing plain language.
- Reading the consent language aloud while the participant follows along and can ask questions.
- Asking the participant follow up questions about the information in the consent document.
- Offering a video or PowerPoint presentation about the study that the participant can watch and listen to.
Plain Language
The guidance recommends following plain language principles for key information section and the entire consent form. (See https://www.plainlanguage.gov/.)
Plain language principles generally involve a combination of text-based and visual approaches (e.g., pictures and diagrams), including
- organizing information with the most important points first,
- breaking complex information into understandable groups,
- using simple language, and
- defining technical terms.
Note: The guidance suggests using these formatting options (e.g., bulleted lists, two-column format, white space), could be appropriate for the entire consent form, not just the key information section.
Follow up Questions
One way that research team members can assist in facilitating understanding is to ask questions to gauge how much the participant has understood after reviewing the consent document. Research has indicated that comprehension is increased if the team member goes through the consent with the participant and then follows up with questions, such as:
“If you were going to tell a friend what this study was about, what would you say?”
“What are the main things that you will do or will happen to you while you are in this study?”
“What are the risks, or bad things that might happen, if you join this study?”
“What are the benefits, or good things that might happen, if you join this study?”
“What can happen if you decide to join the study, but then change your mind?”
Flexible Approaches & Alternative Formats
The regulatory guidance suggests that researchers may utilize alternate platforms (e.g., video, PowerPoint) to help prospective subjects better comprehend the reasons why they might or might not want to participate in research. The use of different formats allows participants who are not as comfortable reading text to engage with audio and video.
Facilitating comprehension in the document might include:
- Making sure the grade reading level of the consent matches the targeted population.
- Providing charts or other formatting in the document to improve ease of reading.
Reading Level
Information should be presented at a reading level comprehensible to the study population. Explanations should be provided for scientific and medical terms.
An assessment of the needs and characteristics of the prospective subject population, including their age, any relevant medical diagnosis, level of English proficiency, education level, and cognitive abilities, can be helpful in developing consent information that facilitates understanding. Information should be provided in the primary language of a prospective subject with limited English proficiency.
Although not required, one way to evaluate whether the information is presented in a way that facilitates understanding is to have the information reviewed by individuals unfamiliar with the research. This may be particularly helpful for forms translated into additional languages.
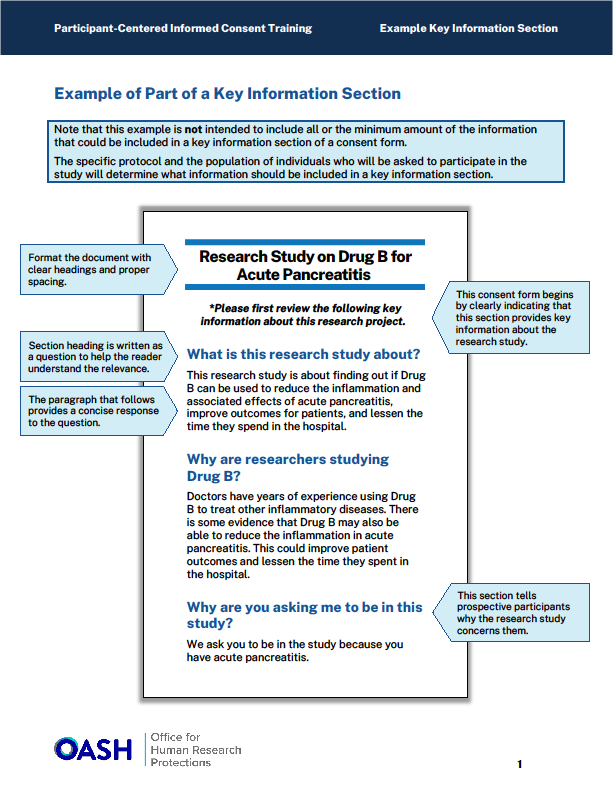
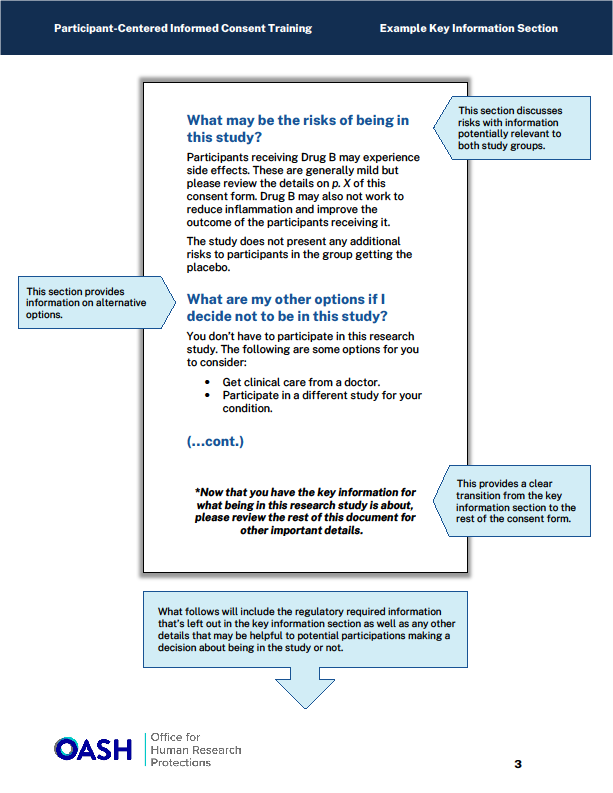
Borders, Columns, Charts and Icons
The guidance recommends organizing information within a defined border (e.g., rounded boxes) creating a discrete unit of information, or including charts to make the content easy to read and understand. Other helpful approaches include formatting text into two columns, and/or using bullet points to simplify long explanations. (See examples in Appendix).
Illustrations or inclusion of icons can also assist the participant with understanding risks or procedures as they read the consent language.
HawkIRB Application Attachments
If using an alternate format to present the key information summary, the IRB must approve what will be presented to subjects in a text version (i.e., transcript of a video).
If utilizing a video,
- provide a transcript on a blank stamped template (available in the dropdown menu in the Consent & Assent Document section) and attach it in the “other consent document” section.
- If the video is available on a study website, include the hyperlink in Section VII.D.16.
- Attach any other alternative key information materials that cannot contain an IRB approval stamp (e.g., PowerPoint presentation, brochure) in the “miscellaneous attachment” category.
Other Resources and Related Research
Appendix
Example #1
Research Study Title: Treatment Consent Key Information Summary DRAFT
Principal Investigator: UI Researcher
This is a summary to help you decide if you want to learn more about being in the research study. The full informed consent document includes more detailed information. Please feel free to ask questions. It is up to you whether you take part in the research. You do not need to take part in this study to receive care for your condition. You can stop taking part in this study at any time without any penalty. | |
| Why are we doing the research?? [Use this space to provide information about the purpose of the research study. Provide enough information so the participant can understand the researcher’s goals. This is a summary and does not need to include the detail that is in the full consent document.] | What will happen, how long will it take, and what will be done at each visit? [Use this space to provide a summary of the procedures that will take place. Provide enough information so the participant can understand what they will be asked to do.] |
| What are the risks? [Describe risks that are the most likely and/or the most serious. This section should be a brief summary, not a duplicate of the risk section of the informed consent document.] | |
| What are the benefits? [Provide an explanation of the specific benefits or state that there are none.] | |
| What can you do instead of being in the research? [Include a clear and concise description of alternative procedures or courses of treatment, if any. For clinical studies, consider first informing prospective subjects about care they would likely receive if not involved in the study, then explain how the care they would receive in the study differs from the standard of care. Focus on increasing awareness of alternatives that may vary based on individual values and preferences.] | |
What if I am injured during the research? Indicate whether medical treatments and/or compensation are available if injury occurs as a result of participation. It is especially important to include this information when there are no plans to compensate for costs related to treatment of research-related injuries.] • There is/is not medical care for injury related to being in the study. Conditions for this coverage are described in the consent document. | What are the costs or payments? • You will have the following costs from being in this study: [SUMMARIZE]. Consult with your insurance company about whether you will have any charges for research-related procedures.
[DO NOT INCLUDE IF THERE IS NO REIMBURSEMENT, INCENTIVES OR COMPENSATION FOR STUDY PARTICIPATION.] • You will receive payment/reimbursement for participating in this study. [DESCRIBE]
|
How participating in the research may affect your daily life: Examples: | |
Example #2:

Example #3:

