Before a PI Leaves the UI: Transfer/Departure/Change of Status Forms and Procedures
Extra! Extra!: Offering Extra Credit as Compensation
HawkIRB Updates: Expanded Character Limits (ROC.5 and ROC.6)
Herky Hint: Assessing Capacity to Consent
Recap: March 2023 Hot Topics Lecture
Recap: ClinicalTrials.gov Reporting Requirements and Consequences of Noncompliance
Medical Ethics Advisor Newsletter, March 2023
In the News
Before a PI Leaves the UI: Transfer/Departure/Change of Status Forms and Procedures
By Kelly O'Berry, BS, CIP

The Human Subjects Office (HSO) and the Division of Sponsored Programs (DSP) offer several forms to collect information and assist Principal Investigators (PIs) who are leaving the UI or transferring to a different status (Adjunct or Emeritus appointment). This article is for faculty members whose status is changing. It is also for study coordinators and departmental administrators who work with faculty who are leaving the UI or changing their UI status. There are separate forms and procedures depending on the funding source and status.
Funded Research
Use the PI Transfer/Departure Form when there is a change in status for a PI who has federal or nonfederal funding for research. A change in status includes transferring a grant to a new institution, terminating their position at the UI, requesting a change in PI for a project to stay at the UI, or changing to adjunct, visiting or emeritus appointment for the PI.
The PI can complete the form themselves or someone else can complete it on their behalf. Complete a separate PI Transfer/Departure form for each funded project. This form notifies the following units about the change in status:
- Division of Sponsored Programs (DSP)
- Grant Accounting Office (GAO)
- Human Subjects Office (HSO)
- Institutional Animal Care and Use Committee (IACUC)
- Environmental Health and Safety (EHS)
Information about the PI Transfer/Departure process is located on the DSP website. Completion is mandatory for funded research. The submitter must know the status of all research under the oversight of the departing PI. Initiate the PI Transfer/Departure process at least 90 days prior to the change in position.
Internal or Non-Funded Research
There is a separate Internal/Unfunded PI Transfer Departure Form posted on the DSP website. The PI can complete the form themselves or someone else in the department can complete it on their behalf. The submitter must know the status of all internal or non-funded research under the oversight of the departing PI; only one complete form should be submitted for all research to avoid unnecessary duplication. Submit at least 90 days prior to the change in position.
Human Research Protection (HRPP) Departure Tool
The Human Research Protection Program (HRPP) Departure Tool is for PIs with departmental, internal or no funding for their research. This tool will help PIs and departments complete all necessary steps prior to the departure/transfer. Submission of this checklist is not required.
The HRPP Checklist Departure Checklist can be completed by the PI or by someone knowledgeable about the PI’s research projects. The checklist collects the following information or prompts the following actions:
- Identify the studies affected by the departure/transfer (IRB#s, Protocol #s, NCT #s, Public and in-development records)
- Identify the affected materials (roles that need to be filled, funding contracts, specimen/data transfer, Investigational New Drug (IND) or Investigational Device Exemption (IDE) with the Food and Drug Administration (FDA)
- A reminder to complete the PI Transfer/Departure Form if the PI has funding for research
- Notify other committees that reviewed and approved the research
- Use the Data Use Agreement flowchart if taking data from the UI to a new institution
- Provide new contact information to the PI’s Departmental Executive Officer (DEO), the IRB and other committees and entities that reviewed the research
- Submit the appropriate updates in HawkIRB – either a Project Closure form or a Modification form to change the PI if the project will continue at the UI
- Submit other updates as necessary – to the Food and Drug Administration (FDA), ClinicalTrials.gov, National Institutes of Health (NIH), and other sponsors or funding agencies
Complete this HRPP Departure Checklist at least 30 days prior to an expected departure/transfer OR within 14 days after an unexpected departure. This Checklist is solely for PI/departmental use. It is not submitted to the IRB or any other committee or office.
eResearch (HawkIRB) Application
The HawkIRB system is only available for people who have a valid HawkID. When a PI leaves the UI, they no longer have access to the HawkIRB system and HawkIRB delegates can only complete limited submissions on their behalf. The PI should pull copies of HawkIRB forms and IRB approval memos and complete all actions in HawkIRB prior to their departure/transfer.
Prior to departure, submit a Project Closure form or a Modification form to change the PI. If there are HawkIRB forms pending IRB review, please complete or withdraw them prior to the departure/transfer.
Project Closure Form – If the research will no longer be conducted at the UI, submit a Project Closure form.
Modification Form – If the research will continue at the UI under the leadership of a new PI, submit a Modification form to change the PI and update consent documents, as necessary.
Withdraw a Pending Form – If the form was sent to the PI through HawkIRB Workflow, add a note in the comment field asking for the form to be withdrawn. If the form is with the IRB, send the request for the form to be withdrawn to irb@uiowa.edu. The IRB will route the form back through Workflow so that request is documented in the HawkIRB system.
Research Team Member Changes – Notify the PI to submit HawkIRB Modification forms for projects in which the departing PI is named on the research team.
Faculty Advisor Change - If the departing/transferring PI serves as the Faculty Advisor for a student/trainee researcher, the student/trainee will need to identify a new Faculty Advisor and submit a Modification form to add them to the research team. The new Faculty Advisor does not need to sign a new Assurance Document. Adjunct faculty may not serve as the Faculty Advisor for a student PI.
Additional Information
The primary source of information about the PI Transfer/Departure Form is on the Division of Sponsored Programs website. View a recorded webinar about the PI Transfer/Departure/Change of Status form. Information about the HRPP Departure Checklist is on the Human Subjects Office website.
Extra! Extra!: Offering Extra Credit as Compensation
By Shane D. Soboroff, PhD
Researchers may use extra credit for compensation when recruiting students as subjects, as outlined in UI Investigator’s Guide and IRB Standard Operating Procedures, Section II, Part 7.K.i. and in line with federal regulations. Researchers offering extra credit to students must ensure (1) that students who decline participation have alternative means for obtaining equivalent credit, and (2) that extra credit is not coercive but offered equitably in line with institutional policies.
Best Practices for Offering Extra Credit to Subjects
Extra credit, like any type of compensation, should not unduly influence students to participate or prevent them from feeling free to decline participation in research. Additionally, instructors offering students the opportunity to participate in research must ensure that alternative means exist for obtaining extra credit, and that these alternatives are equivalent. Here are some tips for offering extra credit, adapted from guidance provided from OHRP and the UI IRB Standard Operating Procedures and Investigator Guide:
-

Make extra credit a small percentage of the overall course grade.
- Include language in consent documents explaining that non-participation will not affect their course grade or relationship with an instructor.
- Offer alternatives that are similar in difficulty and time commitment to research participation.
- Avoid grading alternatives to research participation; subjects in research are not graded.
- Don’t penalize students for withdrawing from a study unless there is clear evidence of bad faith on the part of the student or withdrawal is immediate. Explain circumstances for withholding compensation in the informed consent document.
HawkIRB Guidance
Explain the use of extra credit to compensate student subjects in HawkIRB:
- Report use of compensation in Section VII.E.9 and select “Yes” for “Other” types of compensation in Section VII.E.16.
- In VII.E.17, describe this form of compensation in the text box that opens when “Other” was selected for VII.E.16.
- In the open text field in Section VII.E.19, describe how extra credit is awarded, explaining the bulleted items (see figure).

- Describe any alternative extra credit options for those who decline to participate. These alternatives should be related to the subject matter of the class and not to the topic of the research.
Summary
The IRB may approve extra credit as a form of compensation for research participation when it does not unduly influence students to participate and there are comparable alternatives for obtaining extra credit. It is incumbent on researchers to clearly explain to prospective subjects the conditions for obtaining extra credit through study participation. Researchers must also ensure that student grades will not be affected if they decline to participate.
HawkIRB Updates: Expanded Character Limits (ROC.5 and ROC.6)
By Shane D. Soboroff, PhD
University of Iowa Health Care policy requires documentation of participation in certain types of studies in the electronic medical record. This documentation is referred to as the Record of Consent (ROC). At the request of the UI research community, the Human Subjects Office recently expanded the character limits for the following sections of the HawkIRB application:
- ROC.5: Key complications with the study drug or procedures
- ROC.6: Any other information for emergency personnel
The expanded character limit will allow researchers to enter pertinent information required under UI Health Care policy directly into HawkIRB without attaching additional documents for IRB review.
UI Health Care Record of Consent Policy
According to UI Health Care policy, research participation must be documented in the medical record for any study in which a physical interaction occurs, including studies with procedures requiring tests, examinations, and use of clinic facilities. The HawkIRB system pushes responses from the Record of Consent (ROC) and other sections to an Institute for Clinical and Translational Science (ICTS) web service to create or update the Epic Research Study Description. It is important to provide complete responses in HawkIRB so that complete information is pushed to Epic.
Updates to ROC.5 and ROC.6

]Previous character limits in ROC.5 and ROC.6 resulted in rare cases of researchers attaching information answering these items to the “Attachments” section of HawkIRB rather than in the provided text fields. These character limits have been expanded so researchers and coordinators can provide full information on complications or side effects related to treatments and procedures. HawkIRB can then push information to Epic from these fields.
For currently approved projects, researchers will update ROC.5 and ROC.6 in the next Modification form to include all information previously relegated to attachments. Delete related attachments as soon as the complete description is included in ROC.5 and ROC.6.
For New Project forms, provide a complete response in ROC.5 and ROC.6. The IRB will not accept separate attachments for information that should be provided in response to the ROC questions.
Summary
Expanding character limits for ROC.5 and ROC.6 will allow PIs and study coordinators to provide detailed information to the EPIC system for use in clinical settings. Additional information is available in the Record of Consent FAQ on the Human Subjects office website. The Human Subjects Office will continue to provide updates when changes to HawkIRB are rolled out.
Herky Hint: Assessing Capacity to Consent
By Shane D. Soboroff, PhD
The principle of respect for persons forms the bedrock for informed consent requirements in federal regulations. All researchers, regardless of subject population, are responsible for assessing “capacity to consent”—determining whether a potential research subject understands information provided during the consent process. Assessing capacity to consent applies in all research, even with subjects without cognitive impairments or whose capacity to consent is unlikely to change. This article addresses how to describe procedures for assessing capacity to consent in HawkIRB.
Previous Guidance
This article discusses how to plan for assessing capacity to consent and where to describe these procedures in the HawkIRB application.
Section 7.M.ii of the University of Iowa IRB Standard Operating Procedures and Investigator’s Guide states:
“Individual's capacities, impairments, and needs must be considered in order to develop practical and ethical approaches to enable them to participate in research. A clear understanding of the implications of various cognitive impairments, along with a careful consideration of proposed clinical research methodology, is required. A key factor in participants' decision making is their appreciation of how the risks, benefits, and alternatives to participation in the study apply to them personally.”
Inclusion of Subjects with Limited or Varying Capacity to Consent
Researchers must consider impairments subjects may have and whether these impairments affect a subject’s capacity to consent either at the time of initial consent procedures or over the course of a study. HawkIRB Section VI requires research teams to specify the eligibility criteria for subjects they intend to recruit and explain whether subject populations may include individuals whose capacity to consent may be limited or vary during a study.

A subject’s capacity to consent may be limited due to cognitive limitations, disability, illness, injury, or other medical, social, or psychological factors. Researchers may intend to recruit individuals with limitations to their capacity to consent, or eligibility criteria may not exclude these individuals. Consider a study of vocational services for adults with developmental disabilities. This could include both an adult whose capacity to consent is limited as well as caregivers or job coaches whose capacities are not limited. Answer “Yes” to Section VI.28 if members of the research team are likely to encounter individuals whose capacity to consent may be limited or change throughout the course of a study. If capacity to consent is not an inclusion criterion, and individuals with limited capacity to consent will be enrolled in the study, “Yes” should also be selected for VI.30. Researchers will then need to explain how they will assess a person’s ability to give legally effective informed consent VI.31.

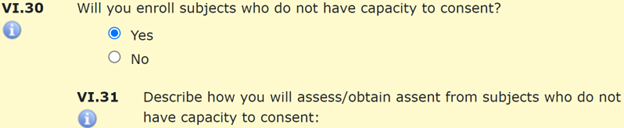
Informed Consent is a process that continues throughout a multi-visit research study. At subsequent study visits after enrollment, researchers confirm that subjects still agree to participate. The IRB asks researchers to consider whether the capacity of a subject to consent to study procedures may change over time. For example, an individual who consents to a study prior to the onset of dementia may require an assessment of their capacity to consent should dementia symptoms advance. Similarly, an individual whose legally authorized representative consents to their enrollment in a study while the subject is in a coma would require assessment of their capacity to consent should they regain consciousness. If the capacity to consent may change over the course of the study, select “Yes” and describe in Section VI.33 how the research team will evaluate the ability of enrolled subjects to give ongoing consent throughout the study.

When subjects are unable to give legally effective informed consent, they may still be able to give assent. In Section VI.34, researchers provide a plan for evaluating ongoing assent from individuals whose capacity to consent might change.
Tools to Assess Capacity to Consent
There are a variety of ways to assess capacity for a subject to provide legally effective informed consent. The Human Subjects Office offers a form for this purpose, “Evaluation to Sign an Informed Consent Document for Research”. If using this tool, download it from the HSO website and explain its use in your New Project Form, following the directions provided above. Add the form as an attachment to the “Other Consent Document” section on the attachments page on HawkIRB. Revise the form where necessary to reflect information specific to your study and/or subject population.
Summary
Informed consent procedures are central to respecting persons in human subjects research. Guidance documents on the HSO website and the “Evaluation to Sign an Informed Consent Document for Research” can help assess a subject’s capacity to consent. For further information, contact us at irb@uiowa.edu or attend office hours to speak with an HSO staff member.

Recap: March 2023 Hot Topics Lecture
By Kelly O’Berry, BS, CIP
On March 22, 2023, the Human Subjects Office gave the tri-annual Hot Topics in Human Subjects Research lecture. This article gives an overview of the information shared in this lecture. The recording is now available in the IRB ICON Course for Researchers.
HawkIRB Updates

There were two recent HawkIRB updates that were announced in the January 2023 IRB Connection Newsletter:
- IRB stamped documents can now be attached as rich text format (.rtf) or Word (.docx) documents. Remember to keep the file format as is – either the way it is on an approved application or the way it is in a template document you generate on the Attachment page.
- New questions about use of Protected Health Information (Section IV.11a and Section VII.D.1)
A few more HawkIRB changes are coming soon:
- A Closure Form memo that can be shared with a sponsor
- New questions about use of the Iowa Clinical Trials Management System (I-CTMS, Section V.27-28)
Human Subjects Office staff are actively working on other revisions and improvements to the HawkIRB system. Read the IRB Connection Newsletter and/or come to future Hot Topics lectures to learn about these changes.
Exempt Form Evaluation
In January 2022, the Human Subjects Office rolled out major updates to the HawkIRB New Project form for research qualifying for Exempt Status. In January 2023, we conducted a Quality Assurance/Quality Improvement (QA/QI) project to assess user experience and satisfaction with the new form. The metrics for Exempt research at the UI show that:
- 7.8% of current IRB approved projects are approved under an exemption category – 4.2% for IRB-01 (Biomedical research) and 23.6% for IRB-02 (Social Behavioral and Educational research)
- In the first year of the revised Exempt form, there was a 38% decrease in processing time for IRB-02.
217 researchers who submitted an Exempt Research application in the past year were surveyed and we received 81 completed surveys (a 37% response rate). Most respondents (75%) were Principal Investigators, but we also received responses from HawkIRB Delegates who completed the survey on behalf of a PI (14%) and some people indicated they were both PI and HawkIRB Delegate (11%).
The majority of respondents (79%) were extremely or somewhat satisfied with the new Exempt Research form. They indicated that the form was:
- Time-saving (78%)
- Easy to use (81%)
- Clearly organized (79%)
- Appropriate (73%)
Respondents were largely positive (52%) or neutral (45%) about whether the application added to the protection of human subjects for their study.
A little over half of respondents (62%) had completed a New Project form for Exempt Research in the past. The majority of those (82%) who had experience submitting Exempt Research in the past felt the new form was much better or somewhat better.
Documentation of Informed Consent and Record Keeping
The federal regulations for the protection of human subjects and UI IRB policy require storage of the signed copy of the IRB-approved Informed Consent Document in the research records (unless the study has a waiver of documentation of consent). The research team must also provide a copy of the Informed Consent Document to subjects (a signed copy if the document contains the HIPAA privacy section).
| Common consent documentation issues | Best practices |
|---|---|
| Out-of-date document | Print documents directly from the Attachments tab of the Project Summary page in HawkIRB |
| Missing pages / missing IRB approval stamp | Store all pages on the signed consent document in the research files. Make sure there is an IRB approval stamp in the upper right corner of every page. |
| Missing information in signature block for subject or Legally Authorized Representative (LAR) and/or the signature block of the research team member who obtains consent | Check for complete signature section at the time of consent – including complete dates (MO/DAY/YEAR), consistent date formatting, content in all required fields |
| Incomplete optional agreements | Make sure all fields are completed at the time of consent, including initials, signature, and dates (as required) |
| Correcting errors | Use a single strike mark to make a correction, then initial and date the change |
There are the same requirements for documentation of consent, even if you use a mailed consent process or an electronic consent document (eConsent). See the educational tools on the Education & Training page of the Human Subjects Office website for guidance about Alternatives to In-Person Informed Consent and use of DocuSign for eConsent.
Store signed consent documents according to the confidentiality protections described in Section X of the HawkIRB application. That includes storing all pages of the document and storing them for the appropriate length of time – 3 years minimum, 6 years minimum if the consent document contains the HIPAA privacy section. Signed documents need to be readily available for review by IRB Compliance Monitoring, sponsor monitors, the Food and Drug Administration (FDA) and/or the Office of Human Research Protections (OHRP).
Using the Consent Summary
The federal regulations for the protection of human subjects (45 CFR 46.116) require the presentation of concise and focused information about a study at the beginning of the consent process. This key information should help potential subjects understand the reasons they may or may not want to participate. This information should be organized in a way that facilitates comprehension and understanding.
The summary should be study-specific and brief. It should contain the most important information that the subject population would want to know to decide whether to learn more about a study. The UI IRB provides a General Consent Summary template in the Consent/Assent Document category on the Attachment page in a HawkIRB application. The UI IRB will also accept a summary that is incorporated into the beginning of the full Informed Consent Document, which is common in sponsor consent templates.
Since January 2019, the UI IRB has required a Consent Summary for studies that have an Informed Consent Document longer than 4 pages. More recently, the Consent Summary was added to the list of consent materials in Section VII.D.15 of the HawkIRB application. Best practices for use of the Consent Summary are to:
- Provide the IRB-approved Consent Summary to subjects before they receive the full consent document.
- Allow time for subjects to read the Consent Summary.
- Talk through the main points of the Consent Summary.
- Ask if a potential subject would like to learn more about the study.
- Although there is no signature section, maintain a copy of the Consent Summary in the research files to document that it was used in the consent process.
Meaningful Filenames – Document Naming Conventions
This section of the Hot Topics lecture reviewed information that was provided in the February 2023 IRB Connection Newsletter, “Making it Plain: Meaningful Attachment Names in HawkIRB.” Meaningful filenames allow for more efficient IRB review. It helps reviewers and the IRB find documents and cross reference between the application and the attachments. This will be very important to keep track of relevant versions as HawkIRB will autopush documents into the Iowa Clinical Trials Management System (I-CTMS).
Researchers often include information in the filename to specify the PI name or other study identifier, the type of document, and the date. For example, “PI name. Consent.01.03.2022” or “Questionnaire.Study Topic.01.03.2022”. Including the date can help to make sure you upload the correct version when editing documents.
Remember to maintain the correct file extension (.pdf, .rtf, .docx, etc.)
There are several options for maintaining version control. Researchers can add the date or a version number in the filename, below the document title, or in a footer in the document itself. However, if you assign your own version number, keep in mind that it will not match the version number in HawkIRB. After the IRB accepts tracked changes in a document, it is uploaded as a new version which will be one higher than the version number the research team member adds to the filename. You can also use the IRB approval stamp for version control.
Recap: ClinicalTrials.gov Reporting Requirements and Consequences of Noncompliance
By Fozia Ghafoor, MBBS

In April, the CT.gov Compliance & Education Manager gave a presentation to provide a brief overview to researchers of ClinicalTrials.gov results reporting requirements and how to keep their records in compliance with the federal regulations. The presentation also outlined the consequences of noncompliance with federal regulations for results reporting requirements.
The presentation covered the following topics:
- ClinicalTrials.gov results reporting requirements
- Deadline for submission of results
- Difference between a primary completion date and a study completion date
- Anticipating a delay in results submission – Submit Good Cause Extension Request
- Good cause extension - Generalized List of Situations
- Consequences for failure to comply with results reporting requirements – FDA and NIH Regulations
- How to prevent violations of federal regulations
To access a recording of this presentation, see the IRB ICON Course for Researchers. The University of Iowa policies regarding ClinicalTrials.gov registration and results reporting can be found in the ClinicalTrials.gov Investigator’s Guide on the ClinicalTrials.gov Requirements page found on the HSO website.
Reporting Requirements for Clinical Trials
If a study fulfills the criteria of an Applicable Clinical Trial or NIH Clinical Trial then the responsible party (PI) will not only register a research study with ClinicalTrials.gov, but the researcher is also responsible for keeping that record in compliance with federal regulations for registration and results reporting. For applicable clinical trials (ACTs) that are subject to 42 CFR 11.42 and NIH clinical trials the standard submission deadline for results information is no later than 1 year after the study's Primary Completion Date, as described in 42 CFR 11.44(a) of the Final Rule. The primary completion date is the date that the final participant was examined or received an intervention for the purposes of the final collection of data for the primary outcome.
Good Cause Extensions
If an investigator is anticipating a delay in results submission, the regulations at 42 CFR 11.44(e)(1)(i) permits a responsible party to request an extension of the deadline for submitting clinical trial results information for good cause. The responsible party must submit the extension request via the ClinicalTrials.gov Protocol Registration and Results System prior to the due date for results information submission. The extension request must include:
- Description of the reasons that the responsible party believes constitute good cause to justify an extension, and
- An estimated date on which the results information will be submitted, with sufficient detail to allow for evaluation of the request.
An investigator can submit more than one extension request for the same clinical trial. All extension requests are reviewed by the NIH director. If an extension request is denied by the NIH director, an investigator can submit an appeal for the denial of request. The deadline for submission of an appeal is no later than 30 calendar days after the date that PRS good cause extension denial notification was issued by NIH.
Medical Ethics Advisor Newsletter, March 2023
By Rachel Kinker, MPA

Medical Ethics Advisor (a publication of Relias, LLC) is a monthly newsletter with articles about human subjects research and medical ethics. Current and past issues of Medical Ethics Advisor and IRB Advisor are posted in the “IRB ICON Course for Researchers.” The portal to this ICON Course is on the Education and Training page of the Human Subjects Office website.
Articles in the March 2023 Issue:
- More Transparency Might Bolster Trust in FDA Advisory Committees
- Out of Options: When Parents Abandon Pediatric Psychiatric Patients at Hospital
- How to Respond to a Consult Request for ‘Difficult’ Family
- Ethicists Can Intervene if Patient/Physician Relationship Is Beyond Repair
- Physicians’ Well-Being Top Ethics Issue
- Efforts Underway to Diversity Clinical Ethics Field
- Should Ethicists Hide Consult Notes from Patients?
- Making More Protected Time for Clinical Ethics Work
- Ethical Problems with Rural Cancer Patients’ Access to Care
- Remote Mental Healthcare Facing Ethical, Legal Pushback
- Physicians Should Educate Patients About Cannabis-Impaired Driving

In the News, April 2023

- Chat GPT in the Clinic? Medical AI Needs Ethicists, The Hastings Center
- National Academies calls for transforming use of racial and ethnic labels in genetics research, Stat
- One way to speed up clinical trials: Skip right to the data with electronic medical records, The Conversation
- How psychedelic drugs may help with depression, NIH