Course-Related Student Projects: The Exception to the Rule
Learning Opportunity: Human Subjects Research, IRB Review Process, and HawkIRB
Increased Civil Monetary Penalties for ClinicalTrials.gov Record Noncompliance
Herky Hints: HawkIRB Filters: A Handy Tool for Research Team Members
Hot Topics and Updates from the IRB and Human Research Protection Program
Division of Sponsored Programs Rebroadcast: Implementation Details for NIH Data Management and Sharing Policy
Write Winning Grant Proposals Seminar and Workshop (2022-2023) – Registration open!
Medical Ethics Advisor Newsletter, July 2022
In the News
Course-Related Student Projects: The Exception to the Rule
By Kelly O’Berry, BS, CIP
Research methods course instructors should all know about one important exception to the rule that research involving human participants requires IRB approval. UI IRB policy has a carve out for projects conducted as a course requirement. Research methods courses often include individual or small-group research projects as a course assignment. IRB approval may not be required when the project satisfies certain requirements and data are not used for any purpose other than the class assignment.
Policy and Checklist

The Course-Related Student Project policy (UI IRB Standard Operating Procedures and Researcher Guide, Section I, Part 12.D) specifies the parameters for class projects that can be conducted without IRB approval. There is a related checklist to help students and their instructors know when IRB approval might be required. It depends on the intent or purpose of the project, the design and procedures, the subject population and how data from the project will be used.
According to the UI Operations Manual and IRB policies, all human subjects research conducted by University of Iowa faculty, staff or students must have approval from the Institutional Review Board (IRB) prior to initiation. This is required if the project is intended to “develop or contribute to generalizable knowledge,” including thesis or dissertation projects. However, research methods course projects are generally more limited in scope and are intended to help students learn how to conduct research. These projects satisfy curriculum requirements and are not intended to further scientific knowledge in a particular field or discipline.
Honor’s and Master’s thesis or Doctoral dissertation projects that meet the regulatory definition of human subjects research will always require IRB review and approval. If a course-related project will be used to support a thesis or dissertation project, then the student researcher should obtain IRB approval prior to conducting the project.
IRB Approval Not Required
Course-related research activities would not require IRB approval if:
- The purpose of the assignment is to teach research methodology.
- The results of the assignment will not “contribute to generalizable knowledge” because they are either being to satisfy a course requirement, or because of limits on who will have access to the results of the project.
- The procedures will be limited to surveys, questionnaires, interview procedures, observation of public behavior, or standard educational exercises.
- The participants will not include prisoners, children, or data about them.
- Data will be recorded without any identifying information (such as code numbers, birth dates, etc.). Or the data are not sensitive in nature, do not pose a risk of harm to the participants’ reputation, employment, financial standing, or do not put them at risk for criminal or civil liability.
- There is no monetary compensation or direct financial support for the project from an external company, organization, or agency.
- The project will not be conducted at the Veteran’s Administration Health Care System (VAHCS) or using any VA resources.
- The project is not conducted or supported by a federal department or agency that follows the federal regulations for the protection of human subjects (the ‘Common Rule’).
The Course-Related Student Projects Checklist is a fillable pdf that students complete and instructors use to determine whether a project qualifies as a course-related student project. The form has pop-up messages if any aspect of the project design indicates that IRB approval might be required. The student is then directed to submit a Human Subjects Research Determination form in the HawkIRB system to ask if the project needs IRB approval.
Information for Participants
Even when a course-related student project can be conducted without IRB approval, the student should share the following information with potential subjects:
- Student name and name of the course
- Course instructor name and their contact information
- Who will have access to individual and summarized results (e.g. instructor, group/team members, the whole class, an outside company/agency/organization)
- That participation is voluntary, and they may stop participating at any time.
Guidance and IRB Overview Presentation
For additional guidance, see the Course-Related Student Project Educational Tool. We can provide an IRB overview presentation about human subjects research:
- What does and does not need IRB approval
- Research ethics
- Criteria for IRB approval
- How to ask if you need approval (HSRD form)
- IRB review process
- Other topics upon request
We invite research methods course instructors to contact the IRB Education & Outreach Program for guidance about course-related student projects or to request a class presentation.
Learning Opportunity: Human Subjects Research, IRB Review Process, and HawkIRB
By Rachel Kinker, MPA
Do you work with or teach individuals or groups that are new to the University of Iowa or learning to conduct human subjects research? Did you know that the IRB Education & Outreach Program offers presentations on human subjects research and the IRB review process? These presentations can be given to research methods courses, journal clubs, research teams, and other groups. Read on to learn how the HSO can help you!

We can give IRB overview presentations that provide a broad overview of topics relevant for anyone who plans to conduct research themselves or work on a research team. This presentation can cover some, or all, of the following topics:
- Guidelines for human subjects research: why and when IRB approval is required
- Basic ethical principles for the conduct of human subjects research
- Student Principal Investigator (PI) training requirements
- What to expect from the IRB review process
- Research off campus or outside the United States
- Course-related student projects
Please Note: We can tailor the presentation to include an orientation to HawkIRB, focus on specific aspects of research activities that are unique to the group, or just offer an open question and answer session about the IRB and human subjects research. We will attend in person or virtually – however the group normally meets.
We recently sent out emails to instructors of research-oriented courses for Fall 2022, offering to give an IRB-related presentation. If you would like an IRB presentation for your class or group, please contact us at irb-outreach@uiowa.edu.
Increased Civil Monetary Penalties for ClinicalTrials.gov Record Noncompliance
By Fozia Ghafoor, MBBS

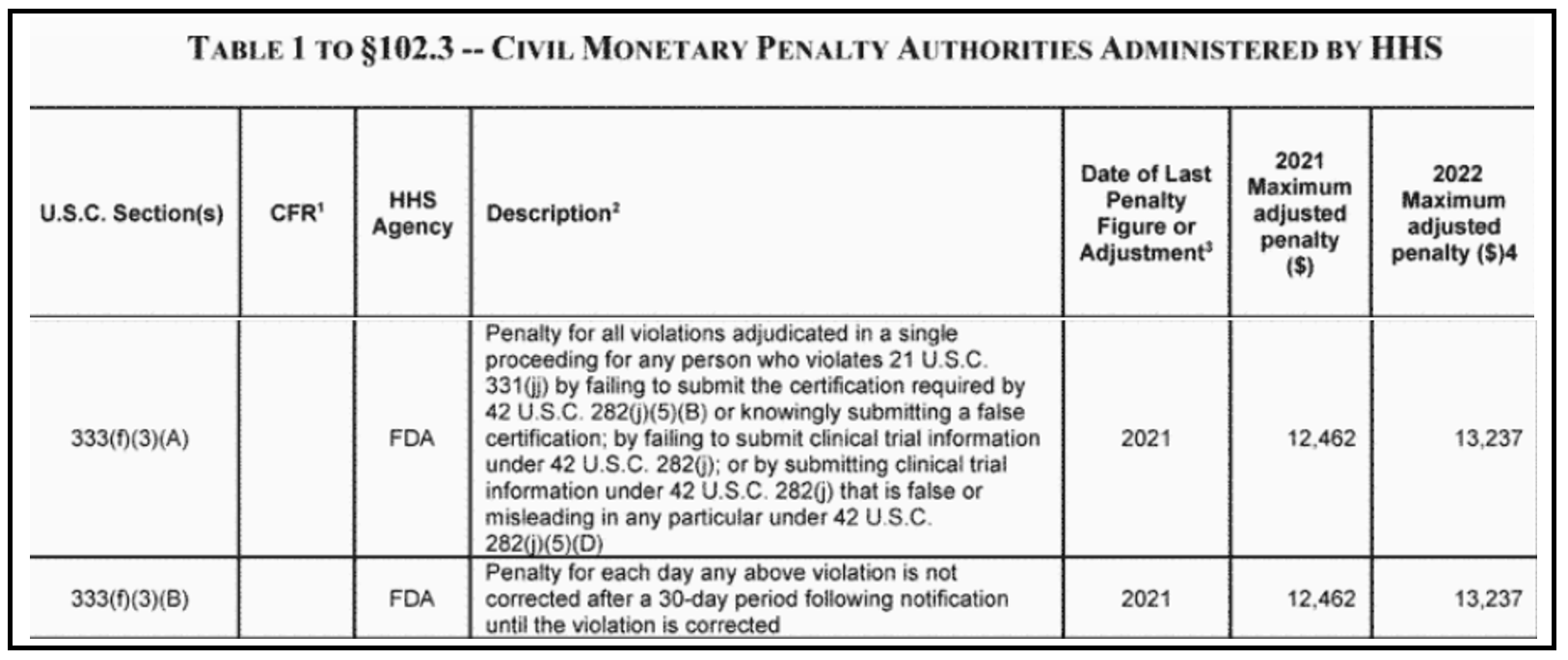
As of July 22, 2022, the U.S. Department of Health and Human Services (HHS) increased the civil monetary penalty for noncompliance with ClinicalTrials.gov registration and reporting requirements. These penalties are adjusted for inflation. For more information, see the Code of Federal Regulations for civil monetary penalties administered by HHS (45 CFR 102.3). The maximum adjusted penalty for ClinicalTrials.gov noncompliance is $13,237, an increase of $775 from the 2021 penalty.
Possible noncompliance includes:
- Failure to register a study on ClinicalTrials.gov
- Providing inaccurate or misleading information for the ClinicalTrials.gov record
- Failure to submit results information by the deadline (1 year after the primary completion date of the study)
The penalty for noncompliance is only applied if the study fulfills the criteria of an Applicable Clinical Trial. If an investigator receives an FDA preliminary noncompliance notice or a warning letter for noncompliance, contact the PRS Administrator as soon as possible. These FDA notices of noncompliance are sent only to the investigator.
(Adapted Penalty Adjustment table from 45 CFR 102.3)
Questions?
Should you have any questions or need assistance with the registration of a clinical trial or submission of results, please contact the PRS Administrator by email: ct-gov@uiowa.edu or phone: (319) 335-6564.
Herky Hints: HawkIRB Filters: A Handy Tool for Research Team Members
By Rachel Kinker, MPA
Research team members need to be able to access a project in HawkIRB in order to conduct the study as it has been approved and to access IRB approved consent documents and other materials. There are filters in the Projects section of the HawkIRB inbox that allow team members to access applications after IRB approval. Principal Investigators (PIs) and study coordinators should work with all team members to demonstrate use of filters to access the HawkIRB application.
Hawk IRB Inbox and Filters
When you login to HawkIRB, you are automatically taken to the inbox with sections for workflow communication (Inbox), Draft Forms, Pending Forms and IRB-approved Projects. Above the Projects section there is a filter with two drop down menus.
The first drop down menu filters by role. Use this filter to see projects that you are associated with as a ‘research team member’ or ‘principal investigator’. The default filter is ‘principal investigator’. If you are a team member and the filter is set to the default ‘principal investigator,’ you will not see the projects that you are associated with as a research team member.
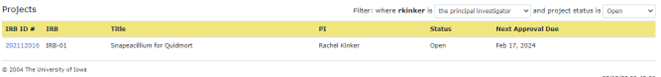
The second drop down menu filters by project status: ‘Any’, ‘Pending’, ‘Open’, ‘Closed’, ‘Withdrawn’, ‘Disapproved’ or ‘Not HSR’. The default filter is ‘Open’. You must use this filter to view projects with any status other than ‘Open’.
Prior to IRB Approval
Prior to submission, only the submitting PI and their HawkIRB Delegates (logged in as the PI) can see a draft form. Research team members can only view the New Project form or subsequent submissions after they are approved by the IRB.

Once the project/form is approved by the IRB, you will be able to see the application simply by adjusting the filter.

Hot Topics and Updates from the IRB and Human Research Protection Program
By Kelly O’Berry, BS, CIP

On July 19, 2022, the Human Subjects Office gave the triannual lecture about hot topics and updates for human subjects research and the Human Research Protection Program (HRPP). Below are highlights from this lecture and additional updates. If you would like additional information about any of these topics, you can view the recording in the IRB ICON Course for Researchers (under Additional Topics).
NIH Research Data Management and Sharing Policy
The new NIH policy replaces the 2003 data sharing policy and is intended to foster data stewardship, advance rigorous and reproducible research and promote public trust in research. Researchers will submit a plan (two pages or less) to the NIH Institute/Center or Office funding the award that specifies how, where and when data will be managed and shared. Plans will undergo peer review with comment, but not a score, on the budget. NIH program staff will assess data management and sharing plans and may recommend revisions. The final plan will be incorporated into the terms and conditions of the award. Compliance will be monitored at regular reporting intervals and may factor into future funding decisions.
The data management and sharing plan must be consistent with the data security plan described in Section X of the HawkIRB application. It must also be consistent with the ClinicalTrials.gov record (when registration is required) and with the information about what will happen to data in the future that is presented to research subjects in the Informed Consent Document.
For the last several months, UI working group has been collaborating to prepare for the new NIH Data Management and Sharing policy that takes effect on January 25, 2023. The working group has representatives from:
- Office of the Vice President for Research – Division of Sponsored Programs and the Human Subjects Office
- Office of the General Counsel
- UI Libraries – Research Data Services
- IT Research Services
- UI Health Care Data Governance Task Force
- IT Security and Policy
The working group is presenting a webinar about this new policy on Tuesday, August 30 from 1:15-2:00 pm (Registration Link) for the UI offices that can assist with implementation answer questions from the UI research community. A recording of this presentation will be posted on the Division of Sponsored Programs website.
For additional information:
- Division of Sponsored Programs webpage about NIH Data Management and Sharing
- For proposal submission questions, the Division of Sponsored Programs has a dedicated email address (nih@uiowa.edu)
- For assistance with creating data management plans, contact UI Libraries, Research Data Services (lib-data@uiowa.edu)
- NIH guidance on Writing a Data Management & Sharing Plan
- NIH two-part webinar series: A Conversation with NIH: Implementing the New Data Management and Sharing Policy
- National Library of Medicine NIH Data Management and Sharing Requirements webinars on demand
Docusign for Assurance Documents
The IRB typically requires a “wet signature” (with a pen) on the Assurance Document attached to the HawkIRB New Project form. In the March 2022 Hot Topics lecture, the Human Subjects Office rolled out several new and updated educational tools. One of them provides a new option for researchers to use DocuSign to obtain electronic signatures on Informed Consent Documents for non-FDA regulated research. In the July 2022 Hot Topics lecture, we advised the research community about the option to use DocuSign for electronic signatures on the Assurance Document.
The Assurance Document is signed by the Principal Investigator (PI), Departmental Executive Officer (DEO) and Faculty Advisor (if the project has a student PI). The IRB expects the PI, DEO and Faculty Advisor to review the New Project form prior to signing the Assurance Document. Each of these individuals makes certain attestations and agreements with the IRB about the following:
- Quality and nature of the application
- Conduct of the study
- Qualifications of the PI
- Scientific merit of the project
- Faculty Advisor guidance and oversight for student PI
Since the start of the pandemic, the IRB has accepted electronic signatures on Assurance Documents if they are obtained through a mechanism that authenticates the signature. The IRB needs to verify that the appropriate persons signed the document. DocuSign provides this authentication. It also has the option to include additional documents for review, such as the printer-friendly version of the application.
For additional information, see the DocuSign Educational Tool. Contact UI Purchasing, Accounts Payable & Travel, for additional guidance on the set up and use of DocuSign (ui-docusign-support@uiowa.edu). Coming Soon – a new educational tool about the use of DocuSign for Assurance Documents.
New Quality Assurance/Quality Improvement (QA/QI) Initiatives
The Human Subjects Office is preparing to launch two initiatives in the Fall of 2022 to evaluate the HawkIRB New Project form for Exempt Research and givee subjects a mechanism to provide feedback to the IRB about their research experience. Watch for additional information later this Fall about these two QA/QI initiatives:
Evaluation of HawkIRB Exempt Application form
In January 2022, the Human Subjects Office (HSO) released a major update to the HawkIRB system for research that qualifies for Exempt Status. The HSO is currently creating a two-pronged evaluation of the new HawkIRB Exempt Application form. There will be separate questions for researchers who completed both the old and the new Exempt application forms and another survey for researchers who only completed the new Exempt application form. The HSO will send survey links to the Principal Investigator and to their HawkIRB Delegate (if the form was submitted by someone other than the PI).
If you submitted a HawkIRB Exempt Application form in the past year, we would very much appreciate your feedback. If you receive an email with the survey link, please take the time to fill out the five-minute survey. Your feedback will help us improve the Exempt Application form and the IRB review process for Exempt research.
Research Subject Feedback
To receive anonymous feedback from subjects with regard to the informed consent conversation and their experience participating in research, the HSO will add a QR code to the IRB Informed Consent Document template. Responses from subjects will help the HSO provide additional guidance to the UI research community to improve the overall conduct of the consent process.
Additional Topics
See the July 2022 IRB Connection Newsletter for articles about the other topics presented during the July Hot Topics lecture:
- Continuing Medical Education Credit for Compliance Monitoring Visits for Biomedical Research
- Tour of the IRB ICON Course for Researchers
Division of Sponsored Programs Rebroadcast: Implementation Details for NIH Data Management and Sharing Policy
The NIH has issued notice NOT-OD-22-189 to inform the extramural research community of implementation details for the NIH Policy for Data Management and Sharing (DMS Policy).
Applicability

The DMS Policy applies to all NIH research, funded or conducted in whole or in part by NIH, that results in the generation of scientific data. The DMS Policy does not apply to research and other activities that do not generate scientific data, for example: research training, fellowships, infrastructure development, and non-research activities. See Research Covered Under the Data Management & Sharing Policy for more details.
Implementation Update
Beginning with receipt dates on or after January 25, 2023, competing applications submitted for activities subject to the DMS Policy will be required to submit a DMS Plan at the time of application for NIH grants or cooperative agreements. The following changes will be made to forms to accommodate the new DMS Plan:
- Data sharing plans and genomic data sharing plans will no longer be submitted to the “Resource Sharing Plan(s)” field.
- A new “Other Plan(s)” field will be added to the following FORMS-H forms to collect a single PDF attachment containing the Elements of a DMS Plan, with details on how to prepare this attachment incorporated into the NIH Application Form Instructions by Fall 2022:
- PHS 398 Research Plan
- PHS 398 Career Development Award Supplemental Form
- An optional Data Management and Sharing Plan format page will be provided to assist applicants with the preparation of this attachment. A preview version of this format page is available now. A final fillable version will be available by Fall 2022 and instructions will be incorporated into the NIH Application Form Instructions. Use of this format page is recommended, but DMS Plans generated using other approaches will be accepted.
- The requested direct costs to support the activities proposed in the DMS Plan must be indicated as “Data Management and Sharing Costs” as follows:
- R&R Budget Form: single line item in section F. Other Direct Costs
- PHS 398 Modular Budget Form: as text embedded within the Additional Narrative Justification
- A brief summary of the DMS Plan and a description of the requested Data Management and Sharing Costs must be included within the budget justification attachment, with details on what information to include incorporated into the NIH Application Form Instructions by Fall 2022; refer to NIH Grants Policy Statement Section 7.1 Cost Considerations for additional guidance on determining appropriate costs:
- R&R Budget Form: embedded within the section L. Budget Justification attachment
- PHS 398 Modular Budget Form: embedded within the Additional Narrative Justification
- These changes will be implemented with application form packages identified with a Competition ID of "FORMS-H" and associated application guide instructions. Additional details will be incorporated into the NIH Application Form Instructions by Fall 2022.
Questions regarding this new policy and how to manage the implications for your research? Please refer to the UI NIH Data Management and Sharing web page for links to UI offices that can assist with questions related to the new NIH policy.
Write Winning Grant Proposals Seminar and Workshop (2022-2023) – Registration open!
It is never too early to start strategizing about how to write a winning grant proposal that will get attention and bring in research funding.
A long-respected program that helps faculty and other research team members do just that, the Write Winning Grant Proposals seminar (Phase I), will be held on Thursday, October 6, 2022. The event will be presented by John D. Robertson, Ph.D., a managing member of Grant Writers' Seminars & Workshops. The seminar is a pre-requisite to the Phase II intensive workshop, scheduled in January 2023. Both events are sponsored by the Research Development Office in the Office of the Vice President for Research. The seminar is appropriate for faculty members, postdoctoral researchers and administrative staff who have had some exposure to writing grant applications, either through training / mentoring or personal experience.

Registration for the Phase I seminar is open now through Sunday, September 18. Please note: there are a limited number of college-specific sponsored registrations complimentary to faculty on a first-come, first-served basis. Those not eligible for a complimentary seat may register under ‘General Registration’.
In person and virtual options are available. (Note: the seminar will not be recorded.) The cost is $150, which includes the seminar, workbook, supplemental materials, and lunch for in person participants.
Phase I Seminar
Title: Write Winning Grant Proposals
When: Thursday, October 6, 8:30 AM – 4:30 PM
Where: IMU Second Floor Ballroom (Virtual option available)
Registration Fee: $150 (includes seminar, workbook, supplemental materials, and lunch for in person participants) A limited number of college specific sponsored registrations complimentary to faculty are available on a first-come, first-served basis.
Phase II Workshop (pre-requisite: Phase I Seminar)
When: January 12-13, 2023
Where: TBD
Consultations: The workshop includes 4-6 months of ongoing virtual consultations
Tuition: $4,000/participant (tuition is split equally between the participant's college, department, and the OVPR; workshop is capped at 25 participants)
To attend: Faculty should contact their collegiate associate dean for research no later than Monday, August 29 for approval to attend
For additional details and to register for October’s seminar: https://research.uiowa.edu/university-iowa-grant-writing-seminar-and-workshop-2022-2023
Medical Ethics Advisor Newsletter, July 2022
By Rachel Kinker, MPA

Medical Ethics Advisor (a publication of Relias, LLC) is a monthly newsletter with articles about human subjects research and medical ethics. Current and past issues of Medical Ethics Advisor and IRB Advisor are posted in the “IRB ICON Course for Researchers.” The portal to this ICON Course is on the Education and Training page of the Human Subjects Office website. This month we are spotlighting some articles about human subjects research from the July 2022 Medical Ethics Advisor Newsletters.
Study Recruitment for End-of-Life Research Raises Ethical Questions
Is it ethical to approach someone near the end of life to participate in research on end of life and palliative care? Most patients would like to be approached and given the opportunity to participate. When the patient is not given the opportunity to participate, it can be viewed as paternalistic. There are additional ethical considerations:
- When patients pass away: researchers should be prepared and ready to respond to loved ones and research team members
- Boundaries: approach the relationship with a focus on the ethics of care
- Minimizing burdens: promote inclusion and ease of access
- Informed consent: the process may take longer for participants to consult with family members and loved ones regarding their participation
- Patients may be reliant on their healthcare providers and feel obligated to participate. Work with individuals outside of the care team to recruit and consent.
- Understand institutional and state policies around a legally authorized representative providing consent.
- Minimize the risk of therapeutic misconception for research participants.-Utilize conscience and direct language and confirm patient understanding and comprehension of the consent language.
- Loss of decision-making capacity in terminally ill patients: -develop plans to assess the decision-making capacity throughout the research process
Variation in Ethical Justification of Randomized Clinical Trials
The term equipoise is often used to justify a randomized clinical trial. Is the term being applied with the same meaning? There are several reasons used to justify randomization:
- Uncertainty of the medical community, individual doctor’s uncertainty, patient’s uncertainty
- Patient’s best interest, risk-benefit tradeoff, methodological rigor, social value, informed consent
Considerations for developing randomized clinical trials:
- Is there adequate statistical power?
- Is the comparator the correct one to best advance the science (placebo, established therapy)?
- Is the study design appropriate to advance clinical science (superiority trial, non-inferiority trial)?
Researchers Need a Plan to Monitor Participants’ Mental Health Risks
Clinical researchers are not in the same position as treating clinicians when it comes to addressing the health needs of patients. There is currently limited guidance on where the responsibility lies for responding to sensitive mental health data and indications of suicidality that may be collected during research. Considerations for investigators handling behavioral and mental health data:
- Consider the characteristics of the trial and study population to determine the likelihood and magnitude of risks
- Identify triggers, thresholds, and responsibilities for action in response to risk-signaling data
- Consider appropriate response mechanisms and capabilities, along with data privacy concerns
- Monitor for mental health risk if the concern could undermine the participants’ ability to voluntarily remain in a study
- Consider that a trial intervention (medication, ‘washout’ period) or procedure could put participants at higher risk for mental health concerns
- Develop a plan to monitor participants and intervene when necessary, rather than excluding patients with increased risks of developing signs of mental health issues
Articles in the July 2022 Issue:
- Ethics Knowledge Gaps Exist on Assessing Capacity, Identifying Surrogate Decision-Maker
- Notable Case Puts Spotlight on Ethics of High-Dose Painkillers
- Q&A: Ethicists Can Help Clinicians Manage High-Dose Pain Medication Cases
- Unexpected Events Make Patient’s DNAR Status Complex
- Few Severe Stroke Patients Receive Palliative Care
- Ethical Concerns Persist with Speculative Cell Banking
- Researchers Offer Definition of Teach-Back for Surgical Informed Consent
- Participant Protection Is IRB’s Mission, but Difficult to Measure
- When Machine Learning Tools Are Used to Predict Suicide Risk
- Financial Conflicts of Interest Are Major Contributor to Academic Success

In the News, August 2022

- Research Ethics Roundup: ethics of insect research, patient engagement in clinical trials, and more, Ampersand, The PRIM&R blog
- Valuing Community Advisory Board Members: An Urgent Call Towards Critical Equity, Ampersand, The PRIM&R blog
- The trust-builder: a cancer center director’s try-it-all strategy for breaking the barriers between research and Black patients, STAT
- Audio Interview: Updated Covid-19 Vaccines and a Look at Monkeypox, New England Journal of Medicine
- Illuminating the brain one neuron and synapse at a time-5 essential reads about how researchers are using new tools to map its structure and function, The Conversation
- Pig organs partially revived in dead animals-researchers are stunned, Nature
- Algorithm That Detects Sepsis Cut Deaths by Nearly 20 Percent, Scientific American
- Scientists Revive Human Retinas after Death, Scientific American
- Common viruses by be triggering the onset of Alzheimer’s disease, Science Daily
- Monoclonal antibody prevents malaria in U.S. adults, NIH trial shows, National Institutes of Health
- Diagnosing hidden hearing loss, National Institutes of Health