How does the University of Iowa prepare for AAHRPP Accreditation?
Recordings Available for IRB Efficiency Initiative, January 2024 Roll Out
UI IRB SOP and Researcher Guide and Policy Changes
Short Form Consent Process: Witness Signature is Required
Optimizing Registration and Results Reporting on ClinicalTrials.gov
FDA Guidance Updates
Promoting Global Clinical Research in Children
In the News
How does the University of Iowa prepare for AAHRPP Accreditation?
By Emily Shultz, CIP
Maintaining the Association for the Accreditation of Human Research Protection Programs (AAHRP) accreditation demonstrates ongoing commitment by the institution to uphold the highest standards in research ethics and compliance with federal regulations, guidance, and institutional policy. Because the University of Iowa Human Research Protection Program (HRPP) has already been accredited, every five years the institution prepares an extensive application for re-accreditation process which includes documentation of all applicable policies and procedures and any changes since the previous application.
The application process for AAHRPP accreditation begins more than a year in advance. The UI HRPP was last accredited in 2020. Our application for reaccreditation in 2025 is due in March 2024. We are currently compiling the documentation for the submission. In the past this was a massive paper submission. This year, AAHRPP has an online application process for electronic submission. We anticipate that the site visit will occur in August or September 2024.
After many years of accreditation, the staff and leadership of UI HRPP understand that the process of reviewing and updating policies is not something that only happens twice a decade: part of maintaining accreditation is adopting a cultural mindset which involves continuously re-evaluating the HRPP policies and procedures and to implement improvements as needed.
However, for institutions seeking initial accreditation, the process can be daunting. Prior to accreditation, an organization must make a thorough self-assessment. The entire research ethics apparatus undertakes an evaluation of all the aspects of the HRPP that is currently in place, and makes amendments or alterations, as needed, to improve operations. Before an organization applies for accreditation, this self-evaluation process will utilize the Standards and Elements that make up the core of the AAHRP Evaluation Instrument for Accreditation.
Standards and Elements
The AAHRP Evaluation Instrument is made up of three primary Domains:
- The organization
- The institutional review board or ethics committee
- Researchers and research staff members
Each Domain is divided into a number of required Standards, each of which may include additional Elements. There are a varying number
of Elements in each of the Standards. For example, in section I (The Organization), there are nine separate Standards that must be evaluated and addressed.
Under the Standards and Elements, the evaluation instrument provides descriptions to assist the organization with completing the application. The descriptions will contain:

- Commentary - that explains how to interpret the Element.
- Regulatory and guidance references - that cite the specific criteria supporting the need for the specific Element in human subjects research.
- Required written materials - includes a list of the general “policies and procedures” that the organization will use to define and communicate its practices in relation to the Element. These would likely include:
- standard operating procedures
- policy statements
- checklists, guidelines, and educational materials
- job descriptions
- forms and templates
- strategic plans
- web sites
- charters, by-laws, mission statements, and other forms used to administer the HRPP
In general, the supporting documents will contain policies and/or procedures. A policy will define a strategy, goal, or objective and will lay out the expectations regarding a course of action. A procedure describes the operational steps that are followed to meet policy requirements.
The accreditation application requires descriptions of procedures:
- How key regulatory terms are interpreted
- Actions that are taken
- The title of the person, office, or entity responsible for taking the action
- Timing of actions
The written procedures should provide enough detail to be understandable to individuals within the organization and should reflect actual practice within the organization.
In our next installments about AAHRP Accreditation, we will provide more details about each of the AAHRP Standards and Elements and how the UI HRPP satisfies them.
The information for this article was adapted from the AAHRPP website.
Recordings Available for IRB Efficiency Initiative, January 2024 Roll Out
By Kelly O’Berry, BS, CIP
At the end of January, the Human Subjects Office (HSO) rolled out the first three components of the IRB Efficiency Initiative:

- Withdraw Forms After 60 Days
- Send Required Actions BEFORE the full IRB Meeting Minutes
- HawkIRB Redesign – Section III (Funding / Other Support)
To review the Jan. 29, 2024 changes in their entirety, visit the IRB Efficiency Initiative page on Office of the Vice President for Research website.
The IRB Efficiency Initiative website contains:
- Landing Page - executive summary and project road map
- Project Updates - progress to date on the initiative
- Initiative Timeline - planned milestones in the project
- Information Sessions - dates and link to register for monthly sessions (noon on the fourth Wednesday of each month)
On the “Information Sessions” page, you can also find links to recordings of past information sessions and brief recorded demos of each change that was rolled out. These recordings are also posted in the of the IRB ICON Course for Researchers, in the section called ‘IRB Efficiency Initiative-Recordings and Documents.’
There are many ways to stay informed about new policies, procedures, and HawkIRB enhancements intended to improve the IRB review process.
Follow hyperlinks in the headings below to access recordings.
New Policy: Withdraw Forms After No Response from the PI for 60 Days
The IRB implemented a new policy to give Principal Investigators (PIs) 60 days to respond when a form is routed back through workflow at any point in the IRB review process. The PI will receive the usual 2-week notices from HawkIRB (Day 14, 28 and 42). They will receive a Day 55 notice that there are 5 days left to respond to workflow. On Day 60 they will receive a notice that the form was withdrawn. This policy applies to the following forms:
- New Project (NP)
- Modification (Mod)
- Human Subjects Research Determination (HSRD)
The policy does not affect Continuing Review (CR) or Modification/Continuing Review (Mod/CR) forms.
New Recreate Option
HawkIRB now has a link for the PI or their HawkIRB Delegate to recreate a form that was withdrawn due to PI inactivity. (If it was withdrawn for any other reason, the PI will not have the option to recreate the form.) If the PI would like to continue to pursue the project at a future date when the outstanding requests can be addressed, they will have an opportunity to recreate the form, but only once.
The recreated form will contain the last round of workflow that was not previously addressed in the withdrawn form. It will also incorporate all application responses as displayed when the form was withdrawn. Upon submission, HSO staff will verify that previous workflow questions were addressed and route the form as a new submission through the regular IRB review process.
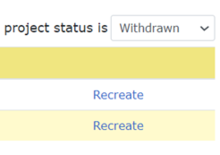
For New Project and HSRD forms:
- Open Project Summary page.
- In “Projects” section, change the project status filter to “Withdrawn.”
- Click the “Recreate” link on the right.

For Modification forms:
- Open the Project Summary page.
- Scroll down to the History section.
- Click the “Recreate link” for the form that was withdrawn.
Address required actions (steps are basically the same for all three form types):
- Click “Final Submission” to access the previous workflow.
- Follow links to address each workflow item.
- Check the box when complete.
- Add comments when necessary.
- Click the blue “Submit Form” box to submit the new form to the IRB.
New Policy and Procedure: Send Required Actions BEFORE the full IRB Meeting Minutes
To give researchers a head start to address required actions after an IRB meeting, HSO staff have the ability to send required actions to the PI prior to receipt of the full meeting minutes. Required actions will generally be sent by the end of the next business day after the IRB meeting. The HSO full board staff will review the required actions with the convened board during the IRB meeting to ensure accuracy. The PI/research team will be notified when required actions are routed for response and when the full IRB meeting minutes are complete.

HawkIRB Redesign: Section III (Funding / Other Support)
To streamline the HawkIRB application and IRB review process, HSO staff worked with Division of Sponsored Programs (DSP) leadership to revise Section III. There are now only three questions in Section III. The first two (Section III.1 and III.2) were revised. We removed questions about type of funding and status of the award. Section III.3, about conflicts of interest in research, remained the same.
Section III.1 contains 10 distinct options for sources of financial funding support or other sources of support for the project for IRB-01 and IRB-02 submissions. IRB-03 submissions have fewer options to correspond with possible VA funding sources. Researchers can select all that apply.
Some external sources of funding/support require linking to an electronic DSP (eDSP) routing form. In Section III.2, there are instructions based on the selections in Section III.1. An attachment category opens in Section III.2 for the PI or HawkIRB Delegate to attach the required documents associated with federal funding sources. This attachment category will also be on the full Attachment page.
UI IRB SOP and Researcher Guide and Policy Changes
By Rachel Kinker, MPA
In conjunction with the January 2024 IRB Efficiency Initiative rollout, there have been changes to the UI IRB Standard Operating Procedures (SOP) and Researcher Guide. A detailed outline of the changes is available on the HSO website. This article provides an overview of the changes.
January 2024 Researcher Guide updates:
Section I, Part 8.A, Surveys of Minors
Documenting a new Iowa state law regarding health and gender related surveys occurring in a school setting with minors. Iowa law now requires parental consent.
Section I, Part 2.J, Clinical Research Unit
The use of the Clinical Research Unit (CRU) requires a partial waiver of HIPPA authorization (see the December 2023 IRB Connection). Utilizing CRU resources and scheduling for CRU services requires access to the potential subjects’ medical record and therefore the research team needs a partial HIPAA waiver.
Section I, Part 9.C.ii, Definition of Continuing Noncompliance
The definition of continuing noncompliance has been revised:

Continuing noncompliance is any noncompliance that occurs repeatedly to the point of suggesting a pattern or underlying problem. Continuing noncompliance may occur due to lack of knowledge (unintentional) or due to deliberate choice to ignore regulations or determinations of the IRB (intentional). Continuing noncompliance could/may include multiple incidents of noncompliance over a period of time or a single ongoing issue over a period of time, which occurs in one or more research protocol(s). OHRP [Office for Human Research Protections] considers the following to be examples of continuing noncompliance:
- The principal investigator (PI) makes the same mistake repeatedly, after the IRB has informed the PI of the problem.
- The PI has multiple problems with noncompliance over a period of time.
- The PI has problems with multiple projects.
The IRB reserves the right to determine serious or continuing noncompliance of circumstances that do not meet this definition. Serious and/or continuing noncompliance is always determined by a convened full board.
Section I, Part 12.F, Quality Assessment/Quality Improvement
Documentation of a UI Health Care requirement to register all Quality Assessment/Quality Improvement (QA/QI) activities in the Clinical Quality, Safety and Performance Improvement Office QI Database.
Section II, Part 11.B.iii.a, Short Form Consent Process
Based on guidance directly from the FDA, if a translator serves as a witness, they must sign the short form consent document (electronically or physically). See also the February 2024 IRB Connection article (Short Form Consent Process: Witness Signature is Required)
References to UI Policy Manual
Updates references throughout the guide, from ‘Operations Manual’ to ‘Policy Manual’
Other Changes
- Removed gender-specific pronouns,
- Updated hyperlinks,
- Grammatical corrections
- HSO staff position titles
- Updates regarding use of an external IRB
Changes Related to the IRB Efficiency Initiative:
We encourage members of the UI research community to view brief recordings and supporting documents about these new policies and procedures in the IRB Efficiency Initiative section of the IRB ICON Course for Researchers.
Section I, Part 11.C, HawkIRB Application Processing/Workflow
New policy regarding withdrawing a project after 60 calendar days of PI\study team inactivity.
Section I, Part 11.C.iv.b, Documentation of Meeting Minutes and Board
Determinations
New policy regarding dissemination of required actions after an IRB meeting, prior to completion of the full meeting minutes.
Short Form Consent Process: Witness Signature is Required
By Kelly O’Berry, BS, CIP

The Human Subjects Office recently received guidance from the Food and Drug Administration (FDA) about the short form consent document and process. The guidance requires that a witness must sign the Short Form Document (physically or electronically). If the translator also serves as a witness, they must be able to sign the document.
The Process
If a research study includes a subject population that does not speak English, this must be described in the HawkIRB application (Section VI.16-17.a), and the IRB must approve the translated Informed Consent Document and other study materials that will be presented to subjects. The research team must do more than just provide translated documents. In order to recruit and enroll non-English speaking participants, the research team must be able to effectively communicate with participants throughout the study. Someone on the research team may be fluent in the language or the research team may hire a translator to translate documents and an interpreter to assist with communications.
However, there are times when researchers are not planning to enroll subjects who do not speak English, but they unexpectedly encounter a potential subject who does not speak English. The short form consent process allows researchers to enroll a limited number of non-English speaking subject even though they did not plan, and obtain IRB approval, to do so.
In the short form consent process, the research team must find someone to verbally translate the IRB-approved Informed Consent Document. Subjects should never be asked to sign a document they cannot read. There must be a witness to the entire consent process who is over 18 years of age, fluent in both languages, not affiliated with the research team able to sign the required documents (physically or electronically). The translator may serve as the witness.
The table below outlines the signatures required on each document:
| Study Documents | Required Signatures |
|---|---|
| Short Form Consent Document |
|
| IRB-approved Consent Document |
|
What’s New?
The new guidance from the FDA, for FDA regulated studies, confirmed that the witness must sign the paper short form and consent documents or use an authenticated electronic signature that is compliant with 21 CFR Part 11 – Electronic Records; Electronic Signatures.
- If the translator serves as the witness, they must be able to sign the document.
- If the translator cannot sign, the research team must find another witness for the consent process.
Researchers at UI Health Care who use Cyracom interpreters to conduct the short form consent process must find a witness in addition to the interpreter. Cyracom interpreters can provide a separate attestation to the consent process, but they are not able to sign documents, physically or electronically.
The short form document has signature lines for the subject, LAR, translator, and witness. The subject or their LAR and the witness will always sign this document. If possible, the translator will also sign the Short Form. When that is not possible, the research team should file the attestation form with the subjects’ other signed consent documents.
Policy and Guidance
The Short Form Consent Process Policy is outlined in the UI IRB Standard Operating Procedures and Researcher Guide, Section II, Part 11.B.iii.a.
For additional guidance about using the short form consent process and documents, see the Short Form Consent Document and Process educational tool.
Optimizing Registration and Results Reporting on ClinicalTrials.gov
By Fozia Ghafoor, MBBS
Recently, the Clinical Trials Transformation Initiative (CTTI) analyzed the underlying issues and challenges with registering and submitting summary results data for applicable clinical trials on ClinicalTrials.gov. This article summarizes the barriers and suggestions outlined in the report for keeping records in compliance with federal regulations.

If a study meets the requirements of being an "Applicable Clinical Trial" (ACT) or receives federal funding for a clinical trial, such as from the NIH, the investigator must register and submit a summary of the results to ClinicalTrials.gov. Certain medical journals, for instance those in the International Committee of Medical Journal Editors (ICMJE), also mandate registration and recommend results reporting on ClinicalTrials.gov. The registration requirement for an applicable clinical trial or NIH-funded clinical study is to be completed within 21 days after enrolling the first participant. Additionally, according to ICMJE guidelines, investigators must register the study even before enrolling the first participant. The deadline for submitting results on ClinicalTrials.gov is no later than one year after the study's primary completion date.
CTTI and the U.S. Food and Drug Administration (FDA) worked together to identify obstacles and challenges associated with trial information submission via the ClinicakTrials.gov database. The objective was to ensure that registration and summary results information for applicable clinical trials on ClinicalTrials.gov is submitted in a timely, accurate, and comprehensive manner. The project included 26 stakeholder interviews and a survey to ascertain and investigate the primary obstacles to the registration and reporting of summary results information for applicable clinical trials, as well as to analyze potential options to address these issues.
The CTTI report outlines common barriers and provides strategies and recommendations for streamlining the process of registration and reporting results.
Here is the summary of the CTTI’s report findings:
- Advantages - The advantages of complete, accurate, and timely reporting on ClinicalTrials.gov for sponsors, patients, and clinical trial investigators include identification of unmet research needs, access to the most recent clinical research, enhanced recruitment options, and the trust of the research community.
- Centralized vs. Decentralized Approach - The comparison of centralized administrative units vs. decentralized approach for managing ClinicalTrials.gov records showed that organizations with centralized administrative units supported teams in adhering to regulations, offered consistency and quality, and aided in a greater success rate and timely submission of results. The drawbacks of a centralized approach include excessive workload for understaffed administrative units, the possibility of miscommunications about timelines, procedures, data, and study protocol revisions, as well as delays caused by waiting for a response from the responsible party (PI).
- Barriers to Registration and Reporting Results - Major challenges in clinical trial registration and summary results reporting include the responsible party's lack of understanding regarding which trials need to be registered or have results reported, uncertainty about when to register trials or submit results for completed or terminated trials, absence of organizational policies on trial registration or penalties for noncompliance, unresponsive principal investigators, lack of concern from investigators about the potential consequences of not complying with federal regulations, poorly defined outcome measures in protocols, lack of harmonization between ClinicalTrials.gov and other regulatory agencies' requirements or registries, unclear organizational policies regarding the responsible party, or when multiple entities are involved, inadequate knowledge about when clinical trial reporting is considered complete, delays in data analysis, and lacking awareness of the publication requirements of journals in relation to ClinicalTrials.gov results submission.
- Tools and Resources - Clinical trials requiring registration and results reporting can be identified through various tools, including internal CTMS tracking and the communication of administrative units with IRBs and the research team.
- Be Proactive - A proactive approach is recommended to address the challenges with registration and summary results reporting. This includes providing information and education to the research team about the consequences of noncompliance with federal regulations and ICMJE policy. Additionally, utilizing a centralized approach, internal tracking, and collaborating with the Institutional Review Board (IRB) can be advantageous.
The University of Iowa employs a decentralized approach in which investigators are accountable for submitting record information on ClinicalTrials.gov. However, the Protocol Registration and Results (PRS) administrator proactively notifies PIs regarding timelines for registration and results reporting. Last year, we developed an escalation process to deal with noncompliant records, which included contacting deans and DEOs in the event of a nonresponsive PI to resolve outstanding issues on ClinicalTrials.gov. The PRS administrator collaborates with IRBstaff regarding studies requiring ClinicalTrials.gov registration and results reporting.
If UI investigators have any questions regarding the ClinicalTrials.gov registration or result submission requirement or the timelines for applicable clinical trials, NIH funding, or ICMJE policy, please contact the UI PRS Administrator at ct-gov@uiowa.edu(link sends e-mail) or (319) 335-6564.
FDA Guidance Update
The FDA recently posted the following guidance documents:
- 2/02/2024 - Conducting Remote Regulatory Assessments Questions and Answers: Draft Guidance for Industry
The Food and Drug Administration (FDA or Agency) is announcing the availability of a draft guidance for industry entitled “Conducting Remote Regulatory Assessments--Question and Answers.” FDA is issuing the draft guidance to describe the Agency’s current thinking regarding its use of remote regulatory assessments (RRAs) in order to increase industry’s understanding of RRAs and facilitate FDA’s process for conducting RRAs. FDA has used RRAs to conduct oversight, mitigate risk, meet critical public health needs and help maximize compliance of FDA-regulated products. This draft guidance provides answers to frequently asked questions regarding what RRAs are, when and why FDA may use them, and how FDA may conduct them, among others.
Chimeric antigen receptor (CAR) T cell products are human gene therapy products in which the T cell specificity is genetically modified to enable recognition of a desired target antigen for therapeutic purposes. This guidance is intended to assist sponsors, including industry and academic sponsors, developing CAR T cell products. The FDA Center for Biologics Evaluation and Research (CBER) Office of Therapeutic Products (OTP) is hosting a virtual public webinar on Thursday, March 7 at 1:00 p.m. to highlight key considerations in the final guidance and address manufacturing, nonclinical, and clinical considerations specific to CAR T cell products. This event is free and open to the public. However, registration is required.
The purpose of this guidance is to provide FDA’s expectations for, and recommendations on, use of a standardized approach for collecting and reporting race and ethnicity data in submissions including information collected and reported from clinical trials and clinical studies2 for FDA-regulated medical products. Using standard terminology for race and ethnicity helps ensure that data are collected and reported consistently in submissions to FDA.
In this guidance, the FDA is providing recommendations to sponsors developing human gene therapy products incorporating genome editing (GE) of human somatic cells. Specifically, this guidance provides recommendations regarding information that should be provided in an Investigational New Drug (IND) application in order to assess the safety and quality of the investigational GE product, as required in Title 21 of the Code of Federal Regulations 312.23 (21 CFR 312.23). The FDA Center for Biologics Evaluation and Research (CBER) Office of Therapeutic Products (OTP) is hosting a virtual public webinar on Thursday, February 29 at 1:00 p.m. to highlight key considerations in the final guidance. This event is free and open to the public. However, registration is required.

Promoting Global Clinical Research in Children
The Multi-Regional Clinical Trials (MRCT) Center of Brigham and Women’s Hospital and Harvard has announced the launch of an updated webpage for their Promoting Global Clinical Research in Children project.
The site offers resources tailored for those who are involved in pediatric clinical research. The resources provided include:
According to the announcement, the Including Young People in Research toolkit provides a number of different tools, checklists, and considerations to assist investigators, Institutional Review Boards (IRBs), sponsors, and others working with children and adolescents to “thoughtfully engage youth and ensure their perspectives are valued, weighted, and integrated.”
The 18 Guiding Principles, provide a framework for the ethical inclusion of children in research. And the series of pediatric-focused webinars include the five-part series Advancing International Pediatric Clinical Research; and the three-part series Prioritizing Young People in Research.
For more information regarding pediatric clinical research visit the MRCT website.
In the News, February 2024
