Herky Hints for HawkIRB: Who's Who? Principal Investigators, Delegates, Team Members, Key Personnel, and Contacts
ClinicalTrials.gov Registration & Results Reporting
By Rachel Kinker, MPA
HawkIRB allows for a variety of ways to manage research team members. The roles and responsibilities of each team member can vary, and the HawkIRB application provides specific areas to indicate each person's role. This article will provide a brief overview of the key roles and responsibilities and where to indicate each of these in HawkIRB.
1. Principal Investigator (PI)
The PI is the lead of the study and is responsible for all aspects of the study, including all research activities conducted by the PI and research team members as well as maintaining compliance with state, federal, institutional and IRB guidelines.
The PI should possess the expertise to conduct the study as described and approved in the HawkIRB application
The PI is responsible for the submission of the New Project form and any subsequent modifications, even if a delegate assists with the entry and submission
All UI PIs must complete the CITI human subjects protection training appropriate for their discipline
Whoever is filling out the application is listed as the PI of the study, unless you are filling it out as a delegate.
2. Student Principal Investigator
The University of Iowa permits graduate and undergraduate students and trainees to serve as the PI of a project.
Student PIs are required to name a faculty advisor (FA) to oversee their research. The student PI must add the advisor to the research team in HawkIRB and as a contact person in Section II.
Student PIs are required to complete two HawkIRB training courses that are offered in the IRB ICON Course for Researchers
Student PIs are required to complete the CITI human subjects protection training appropriate for their discipline
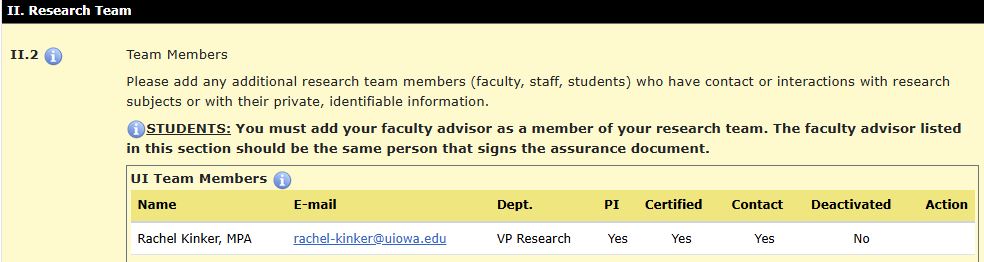
In HawkIRB, Section II, item II.3 the student PI will select their role (graduate/undergraduate) and a link to the “Student Principal Investigator Training” will appear. If the training has been completed, a green “complete” will appear, if not it will show a red “incomplete.”
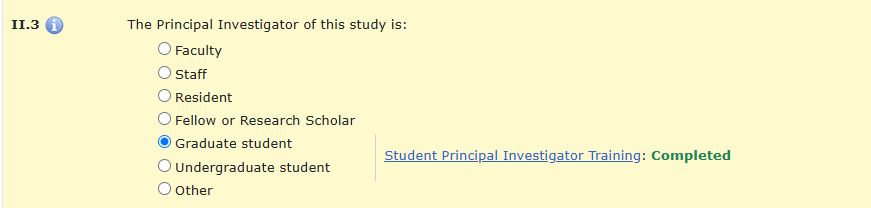
When Grad/Undergrad is selected, item II.3.a will appear with a drop-down menu to select a faculty advisor, the FA must first be added as a research team member in item II.2 for them to appear as a selection in II.3.a
3. Faculty Advisor (for Student PIs)
When the PI is a student, a faculty advisor (FA) must be named to the research team to oversee the research project.
The FA must be a University of Iowa faculty or staff member
The FA must be listed in the HawkIRB application as a research team member for the projects (see above for Student PIs)
When an FA is overseeing a project, they must be able to provide sufficient time, attention, and guidance to the Student PI
The FA is responsible for ensuring the project design is scientifically sound, the application is complete and detailed, and that the study is conducted in an ethical manner
The FA must sign the Assurance Document in HawkIRB, which constitutes the agreement the FA makes with the IRB about how s/he will supervise the student PI’s conduct of the research to protect human subjects.
4. Research Team Members
Anyone that is in direct contact with research subjects during the study procedures and/or has access to subjects’ identifiable private information or biospecimens while working on the research study.
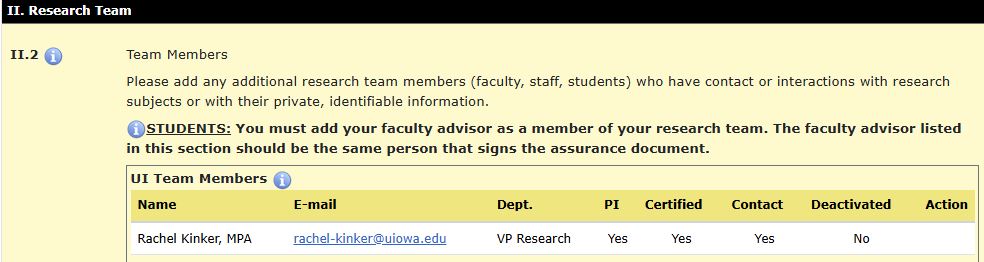
In HawkIRB, all research team members are listed in Section II. Research Team.
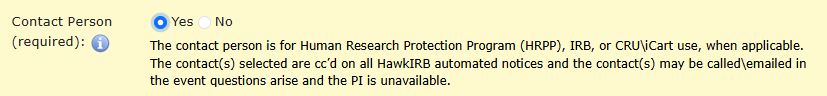
When adding a research team member, you are able to indicate if they are a contact person. Contact persons will receive automated messages from the HawkIRB system regarding the application. Designated contact persons are not able to amend the application unless they are also a delegate for the PI. Anyone listed as a contact person should be able to answer questions about the project from the Human Subjects Office (HSO) staff and the IRB
Anyone that is added as a research team member also must be added to the Certified Investigator database prior to submitting the form. This means that anyone to be added must complete the required CITI human subjects protection training appropriate for their discipline (as well as the Student PI HawkIRB trainings if they are a student PI)
This appears in HawkIRB, Section II. Research Team, item II.2
5. Key Personnel
Team members with decision-making authority such as the PI, co-investigators, and other faculty members are generally considered key personnel.
Individuals who are likely to be authors of manuscripts or present research findings at a national conference would likely be included.
Key personnel would not include administrative personnel or individuals that perform routine, pre-defined, or incidental tasks related to the project.
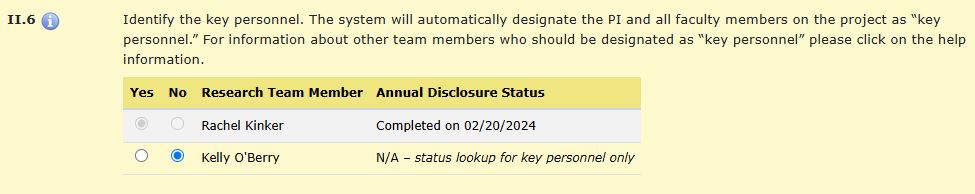
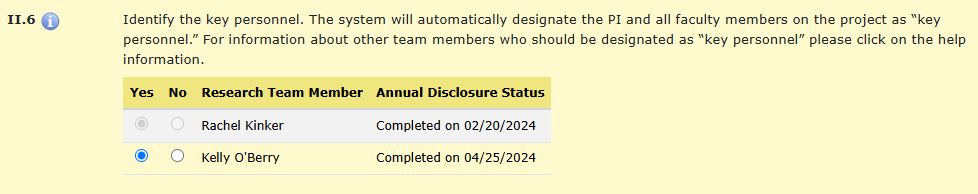
HawkIRB automatically designates the PI and all faculty members on the project as Key Personnel.
Any research team member who is listed as key personnel will have the status of their Annual Disclosure of Outside Activities displayed in HawkIRB. Anyone identified as key personnel must complete a financial interest disclosure form (eCOI) prior to the application being submitted. HawkIRB will not allow the application to be submitted if anyone identified as key personnel has not submitted the financial interest disclosure form. Financial interest disclosure must be submitted annually.
This appears in HawkIRB, Section II. Research Team, item II.6
6. Delegates
The PI can name a delegate in HawkIRB. The delegate can manage submission and respond to workflow on behalf of the PI. A delegate has the same permissions within HawkIRB as the principal investigator.
Only the PI can assign or remove delegate permissions, this is controlled by through the Delegate Permissions System within HawkIRB
For guidance on managing delegates, refer to the HawkIRB Delegates page. This resource provides instructions for adding/removing a delegate and turning on/off the Delegate Permissions System.
7. Research Methods Course Instructors
While research methods course instructors do not have a specific area in HawkIRB (unless they are acting as a faculty advisor), they do have a unique responsibility to guide students in conducting ethical research. Research methods course instructors should be familiar with HawkIRB guidelines to determine if IRB approval is needed for class projects. Additional guidance is provided for Research Methods Course Instructors and Course-Related Student Projects.
Questions about this topic? You can email the HSO Education and Outreach team or come to Office Hours to speak directly to a member of the HSO team.
By Fozia Ghafoor, MBBS, MS
ClinicalTrials.gov serves as a pivotal platform for clinical research studies, ensuring transparency, accountability, and public trust. It provides a publicly accessible repository of information on clinical studies, enabling patients, researchers, and healthcare professionals to stay informed about trial designs, objectives, and outcomes. The principal investigator holds the critical responsibility of registering their research studies and summarizing the trial's outcomes accurately on ClinicalTrials.gov. Adherence to federal regulations under FDAAA 801 and ClinicalTrials.gov review criteria is essential not only for maintaining compliance but also for the integrity of clinical research.
September Presentation Overview
In September, the ClinicalTrials.gov compliance and education manager presented the critical aspects of trial registration and results reporting. The session aimed to demystify the processes, enhance data accuracy, and minimize common errors. The presentation aimed to enhance understanding of federal regulations, institutional policies, and the resources available to researchers.
The presentation covered the following topics:
Overview of the ClinicalTrials.gov database
ClinicalTrials.gov is a public database offering a centralized repository for clinical study information. Its dual purpose includes informing the public and being a critical resource for researchers and healthcare professionals. The National Library of Medicine (NLM) manages the database, conducting a limited review of submitted record information to ensure compliance with federal guidelines.
Which trials need registration and results reporting?
Trials requiring registration include applicable clinical trials (ACTs), NIH-funded studies, and those under WHO, PCORI, ICMJE, Department of Defense (DOD), and Centers for Medicare & Medicaid Services (CMS) guidelines. The registration deadlines generally fall before the enrollment of participants for ICMJE, WHO, DOD and PCORI to within 21 days of the first participant's enrollment for ACT and NIH clinical trials. Results should be reported no later than one year after the study’s primary completion date.
How to register a study, report results, and update records
You can create a Protocol Registration and Results System (PRS) account by emailing the PRS administrator at ct-gov@uiowa.edu. The information in the protocol section is required at the time of record registration, while the document section needs to be completed at the time of results submission.
Protocol Section has 13 subsections, covering details regarding study identification, study status, sponsor/collaborators, oversight, study description, study design, arms and intervention, outcome measures, eligibility criteria, contacts/location information, IPD sharing statement, and references.
Documents Section requires study protocols, statistical analysis plans (SAPs), and informed consent forms, as applicable.
Results Section requires information regarding participant flow, baseline characteristics, outcome measures, and adverse events.
The presentation emphasized timeliness, consistency with the study protocol, and clarity in submitted data as critical factors.
Common errors during registration and results reporting, and how to avoid them: Major errors in 3 main sections of the record include
Protocol Section: Errors such as mismatched protocol IDs, records not verified and released in the last 12 months, vague outcome measures, and missing secondary IDs were highlighted. To avoid these issues, researchers should align their records with IRB-approved protocols and ensure consistent updates.
Documents Section: Missing or improperly formatted documents, omission of required cover pages, or using HawkIRB applications in place of study protocols can lead to compliance issues.
Results Section: Incomplete or poorly presented results, such as inconsistent participant flow, unclear study population at risk in the adverse event section, multiple outcome measures reported together, and unclear time frames for outcomes, are frequent errors. These issues can delay record processing and posting on the ClinicalTrials.gov public site.
Navigating the PRS support system and help content
PRS Support System: Offers section-specific help and definitions. For more detailed information investigators can also find PRS user’s guide, PRS guided tutorials and examples for results data entry under the “Help” tab on the CliniaclTrials.gov home page.
Institutional Guidelines: The ClinicalTrials.gov Investigator’s Guide and HawkACT checklist provide comprehensive compliance guidance tailored to provide information for record registration and results reporting.
Overview of ClinicalTrials.gov PRS Beta website
The PRS beta website was launched in August 2024. It provides enhanced features for protocol registration. However, document and results sections are currently only available on the classic PRS site.
The presentation can be viewed at the IRB ICON Course for Researchers.
For detailed federal and institutional guidelines regarding ClinicalTrials.gov PRS, visit the ClinicalTrials.gov resources section under ClinicalTrials.gov page on the Human Subjects Office website or reach out to the PRS administrator at ct-gov@uiowa.edu.