Adverse Events and Unanticipated Problems
Herky Hints for HawkIRB: Reportable Events
Adverse events (AEs) and unanticipated problems involving risk to subjects or others (UPIRTSOs) are categories of reportable events that may occur during a research study. How they are different, and what reporting requirements are involved, are important distinctions for researchers, members of the Institutional Review Board (IRB), and Human Subjects Office (HSO) staff to understand.
Reportable Events
At the University of Iowa, a reportable event is a broad category that includes any incident that occurs in a research study that needs to be reported to the IRB via HawkIRB. The UI Standard Operating Procedures and Researcher Guide outlines five events that should be submitted on a reportable event form (REF) in HawkIRB:
Serious adverse drug event
Serious adverse device effect
Unanticipated problem involving risk to subjects or others
Receipt of new information (including risk or benefit) that may impact the willingness of subjects to participate or continue participation in the research study
Any incidents of noncompliance with the federal regulations or the requirements or determinations of the IRB
Investigators must report to the appropriate UI IRB if any of the items above occur in a study where IRB-01, IRB-02, or IRB-03 is the IRB of record. For studies approved by an external IRB, report events directly to the IRB of record according to their reporting requirements. (See also the Herky Hints for HawkIRB: Reportable Events.)
Researchers have a responsibility to report an event to the IRB so that the IRB can evaluate the occurrence and decide what actions are needed to:
protect human subjects and
comply with federal regulations and institutional policies.
When the REF is submitted in HawkIRB, an IRB chair will review the REF and compare the content with the approved project application, informed consent document(s), protocols, and investigator brochures to determine whether the event meets the definition of an AE and/or a UPIRTSO. If the chair finds that the event is likely to meet the reporting requirements, the chair will refer the REF to the convened IRB for review.
The requirements for a researcher to communicate a reportable event beyond the UI IRB depends on the specific criteria of an adverse event or an unanticipated problem depends on several factors.
Definitions
According to Office of Human Research Protections (OHRP) Guidance, unanticipated problems include any incident, experience, or outcome that is:
unexpected,
related or possibly related to participation in the research, and
suggests that the research places subjects or others at a greater risk of harm than was previously expected.
Events that were not described in the study protocol, investigator brochure, and/or the informed consent document and pose an increased risk to subjects or others would be reported as a UPIRTSO. The “O” for “others” in UPIRTSO could include a member of the research team, family members of the enrolled participant, or anyone else who faced unexpected and increased risk of harm.
Food and Drug Administration (FDA) guidance defines an adverse event as:
“unexpected, serious, and would have implications for the conduct of the study”
Adverse Drug Events: If the reaction was not described in the study protocol, investigator brochure, and/or informed consent document; is serious in nature; is related to a study drug; and occurred with a subject enrolled at the UI or VA Health Care System (VAHCS), then submit a serious adverse drug event REF.
Adverse Device Effects: For serious events related to a device, submit a serious adverse device effect REF for all events that occur with a UI or VAHCS-enrolled subject. Submit an adverse device effect REF for unanticipated events that occur with a non-UI/VAHCS subject.
Although events could fit into more than one category, the PI decides which one is most appropriate based on these definitions and available guidance. If an IRB chair or the board determines that the event fits better in a different category, they will route the form back for the PI to change the category.
Reporting by the IRB
Health and Human Services (HHS, 45 CFR 46.108(a)(4)) and FDA (21 CFR 56.108(b)) regulations state that the IRB is responsible for prompt reporting of:
Any unanticipated problems involving risks to subjects or others
Any serious or continuing noncompliance
Any suspension or termination of IRB approval
Institutional Reporting Policy
Institutional policies for reporting by the IRB are in the UI Researcher Guide, Section I, 9.C IRB Determinations Requiring Reporting and 9.C.i Suspension or Termination of IRB approval.
In addition to internal notifications to UI research administration, the IRB will also notify the following, as applicable:
Office for Human Research Protections (OHRP)
Food and Drug Administration (FDA)
Other federal funding agencies subject to “The Common Rule,”
UI Division of Sponsored Programs (DSP) - for reporting to external sponsors
UI/VAHCS officials – for studies under IRB-03
Reporting by the Sponsor or Sponsor/Investigator
Typically, the study sponsor takes care of reporting adverse events and UPIRTSOs to the FDA. However, if the UI principal investigator holds the Investigational New Drug (IND) or Investigational Device Exemption (IDE) with the FDA, they are responsible for reporting to the IRB, sponsor, and FDA. The FDA regulations also require that the investigator will:
… report to the IRB all changes in the research activity and all unanticipated problems involving risks to human subjects or others, and will not make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to the human subjects. 21 CFR 312.53(c)(1)(vii)
The UI Researcher Guide (Section II.9.D.iv.a.v) provides information on reporting requirements for the investigator to the sponsor. The sponsor will also have specific reporting requirements. FDA guidance about adverse device effects under § 812.46(b) state that the researcher “shall report the results of such evaluation to FDA and to all reviewing IRB's and participating investigators within 10 working days after the sponsor first receives notice of the effect. Thereafter the sponsor shall submit such additional reports concerning the effect as FDA requests.” (21 CFR 312.32(d))
Summary
Factors such as federal regulations, funding agencies, and institutional policies determine whether a research occurrence is a minor occurrence or is an adverse event, an unanticipated problem, or both. Working together, researchers and the IRB can fulfill their respective roles in protecting research participants and fulfilling reporting requirements.
Questions about this topic? You can email the HSO Education and Outreach team or come to Office Hours to speak directly to a member of the HSO team.
Have an idea for an IRB Connection newsletter article? Let us know!
A reportable event is any problem in a research study that needs to be reported to the IRB, ranging from a small mistake to a more significant issue. Reporting these events ensures that research is conducted ethically, safely, and in compliance with federal regulations. (Note: The HSO has developed a Reportable Event Decision Tree to help guide researchers through the process.)
Personnel must use the reportable event form (REF) in the HawkIRB system to disclose all reportable events to the appropriate UI IRB (for IRB-01, IRB-02, or IRB-03). For studies approved by an external IRB, these events are reportable directly to the IRB of record
The investigator must submit reports via HawkIRB within 10 working days of the event or within 10 working days of the PI becoming aware of the event. For IRB-03 studies, reporting must occur within five working days.
Information Provided
The HawkIRB reportable event form (REF) requests information including:
a description of the event,
the date of occurrence,
whether it is a local or external report,
an explanation of how the event affected the rights, safety, or welfare of the subject or others,
the status of subjects, and
any changes or modifications to the project that the research team plans to make because of the event.
If the REF is an event of noncompliance, a corrective and preventive action (CAPA)
Completing the REF
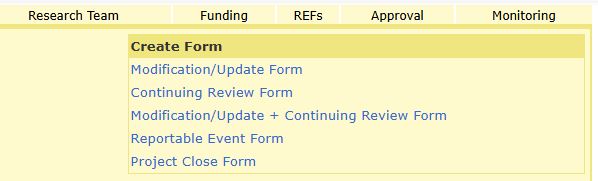
To begin the form, log into HawkIRB and select the applicable project number under the ‘Project’ heading. This will direct you to the project summary page. In the upper right corner, the ‘Create Form’ box lists the form options, select the ‘Reportable Event Form.’
You will then be directed to the REFs tab where you can either update or modify an existing REF or create a new reportable event form by clicking the link circled below.
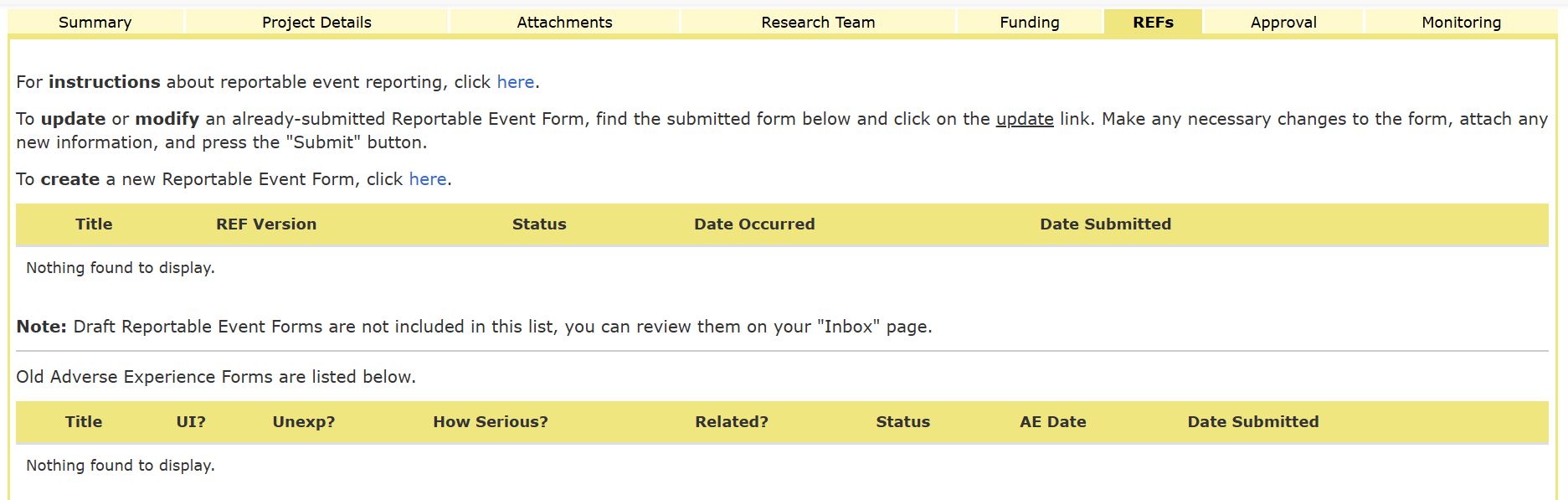
The REF Index will appear. Click on the ‘Reportable Event Type’ to begin completing the REF.

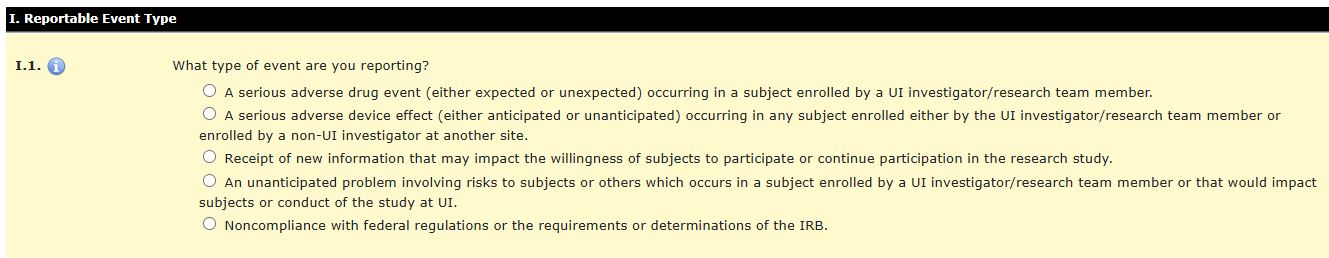
For item I.1, select the type of event you are reporting. The options are:
A serious adverse drug event-if participating in a multi-site study and incident occurs at another site, you would not report here.
A serious adverse device effect-if participating in a multi-site study and incident occurs at another site or at the UI, you would report here
Receipt of new information- if participating in a multi-site study and incident occurs at another site or at the UI, you would report here. This can be positive (early efficacy/benefit) or negative (identify trend that impacts subject safety). This could also include changes in the standard of practice/care or results of new research.
An unanticipated problem-any event or problem
Not expected based on the nature of research, study population, and/or the approved protocol and procedures
Impacts the rights/safety/welfare of subjects or others (research staff or subjects’ family members)
Related to research interventions, procedures, and/or conduct of study
Noncompliance with federal regulations—incident when personnel did not conduct study procedures as described in the approved IRB application and/or in violation of federal regulations for human subjects research
For the selected item you will then provide a name and/or a brief description for the form in item I.2.
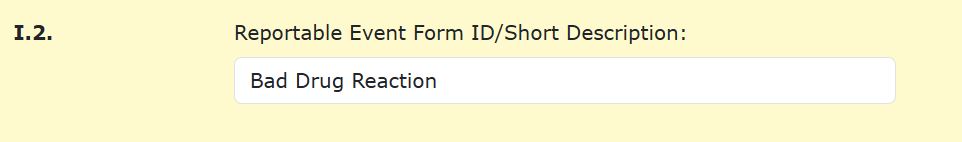
Note that depending on which type of event you select, different questions will open or close different sections of the form.
Describe the event and continue to answer the questions in each section, making sure to select the ‘Continue/Save’ button as you move through the form.
Attachments
In the attachments section you may include anything that you would like to share with the IRB. Do not include any attachments that contain identifying information about participants. Attached documentation might include the researcher’s corrective action plan, which aims to prevent a similar occurrence in the future.
After the REF is Submitted
When the REF has been submitted, the status will change to ‘Pending.’ From there, there are two outcomes: reviewed or withdrawn.
If the form is reviewed, an IRB chair will read it and may refer it to the board for full IRB review, if it meets the reporting requirements. The chair or board will communicate with the researcher about any actions that need to be taken because of the REF.
Note: The IRB does not ‘approve’ REFs; they are simply reviewed and then filed within the HawkIRB system.
If the form is withdrawn, the REF did not meet reporting requirements of the UI IRB, but you will want to keep the information about the event as a part of the total research record, and you may need to submit it at the time of continuing review, if applicable.
Questions about this topic? Email the HSO Education and Outreach team or come to Office Hours to speak directly to a member of the HSO team.
Have an idea for a future Herky Hint for HawkIRB? Let us know!