Index
From the Director
Getting Started: Do I Need IRB Approval?
Herky Hints: Informed Consent Document Checklist
IRB Advisor, January 2018: Right to Try in Oncology: Gatekeepers or Mercenaries?
In the News
From the Director
By Michele Countryman
The eResearch application (HawkIRB) system has been updated to accommodate requests to be removed as a Principal Investigator’s (PI) delegate. This revocation request may be as a result of a change in delegate responsibilities or departments, a PI departure, etc. An assigned delegate can now submit a request for the PI to remove their delegate status to one or more projects they may be assigned to edit on behalf of the PI. The steps to request a revocation of access are posted on the Human Subjects Office website. (These instructions are available via institutional log in with UI HawkID@uiowa.edu and password.)
For more information on setting up the delegate permission system, visit the HawkIRB Delegate Permissions FAQ on the HSO website. For information on who can be a delegate, visit the Delegates in HawkIRB FAQ.
Getting Started: Do I Need IRB Approval?
By Maegan Tyrrell
There is a question that researchers ask Human Subjects Office (HSO) staff and IRB Chairs quite regularly; “I have an idea for a project. Do I need IRB approval for that?” In fact, there is a short form in the HawkIRB system that researchers can use to ask this question. The Human Subjects Research Determination (HSRD) form is designed to collect all of the information an IRB Chair needs answer this question. First the Chair determines if the project meets the regulatory definition of research and then they determine whether human subjects are involved. This article provides these definitions and some information about the HSRD form.
Definition of Research
The regulatory definition of research is “a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.”
- “Systematic investigation” refers to the process of the project. The researcher collects the same information, does the same tests, takes the same measurements, asks the same questions from each subject.
- The intent or purpose of research is to answer a question, or more than one question.
- “Research development, testing and evaluation” refers to pilot studies or a small study that might be the beginning of a larger project.
- “Develop or contribute to generalizable knowledge” means the researcher will draw some conclusions and make generalizations based on the information collected so that the results might be useful to someone else.
Note that the regulatory definition of research is not dependent on whether the results will be published. Lots of research meets this definition but never gets published. The decision on needing IRB approval is based on the study design and the intent or purpose of the project, not what will happen with the data afterwards.
Definition of a Human Subject
The regulations define a human subject as “a living individual about whom an investigator (whether professional or student) conducting research:
- Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
- (ii) Obtains uses, studies, analyzes or generates identifiable private information or identifiable biospecimens.”
Think of an intervention as drawing blood, taking measurements, or giving a treatment. An interaction is any kind of interpersonal communication; questionnaires, surveys, interviews, focus groups, etc.
Most people recognize the first part of this definition, but do not always consider that use of identifiable private information or identifiable biospecimens would require IRB approval when it is done for research purposes. Even if the researcher never has contact with the subject, and the subject doesn’t know the researcher is using their data, it is human subjects research if the information or biospecimens are being used for research purposes. The IRB typically uses the list of 18 HIPAA identifiers to determine if information or biospecimens contain identifiable information.
Using the Human Subjects Research Determination (HSRD) Form
UI faculty, staff and students can complete a HSRD form in the HawkIRB system to ask if a project needs IRB approval. The HSRD form is not required by the IRB. It is a tool to assist researchers in determining if IRB review and approval is necessary.
Generate an HSRD form in the black menu bar in the Principal Investigator’s HawkIRB inbox. This is a very short from (only four sections) that typically goes through the IRB review process very quickly. CITI training is not required to fill out and submit this form. The HSRD form is not the first step in getting IRB approval. If a project meets the definition of human subjects research and definitely needs IRB approval, the researcher should submit a New Project form.
After submission, the HSRD form goes through a brief prescreen to ensure complete responses are provided and then directly to an IRB Chair. HSRD forms are typically reviewed within two to four business days. The Chair will either make a determination or route the HSRD form back with questions or requests for clarifications about the project purpose or procedures. When the IRB Chair makes a determination about whether or not IRB approval is required, there will be a determination memo attached to the HSRD form.
- Project is Not Human Subjects Research - The determination memo will state that as long as the project is conducted exactly as described, there is no need for IRB approval.
- Project is Human Subjects Research - A memo will still be attached to the HSRD form, but what is more important is the draft new project form that will be started in HawkIRB. The new project form is populated with some of the information from the HSRD form. The researcher needs to finish filling out the form and submit it once completed to move forward with the review process.
HSRD form Metrics
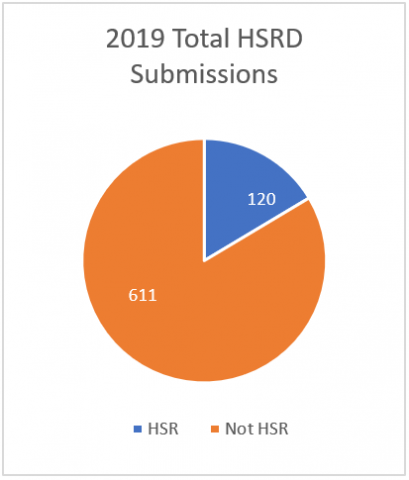
The moral of the story is to always ask if you are not sure whether you need IRB approval. Fill out the brief HSRD form to provide all the information the IRB Chair needs to determine whether IRB approval is necessary. You can save yourself a lot of time by asking up front before filling out an unnecessary New Project form.
Herky Hints: Informed Consent Document Checklist
By Joanie Neyens
A new tool has been created to facilitate consistency between the HawkIRB application and the Informed Consent Document. If you have attended a HawkIRB training session, or if you are a seasoned researcher or study coordinator, you know anytime a HawkIRB application is returned to you in workflow, IRB review of the application stops. The more times this happens, the longer the review process. One of the reasons that applications are returned to the researcher is due to inconsistencies in the information provided in the HawkIRB application and the attached documents—especially the Informed Consent Document. This new Informed Consent Document Checklist can be used to make sure the Informed Consent Document is consistent with the HawkIRB application.
The checklist is divided into two parts:
- Informed Consent Document – General – Includes template sections that are included in most informed consent documents, regardless of the type of study or the study procedures.
- Informed Consent Document – Study Specific – Includes template sections of the consent form that are unique to the study design.
Both sections of the checklist provide specific references to the applicable section/s of the HawkIRB application so you can ensure the information you put in the informed consent document is the same information that you provided in the application. Additionally, both sections provide a key for which sections of the informed consent contain the elements of consent that are required by the federal regulations, UI policies or other regulations (i.e. HIPAA, NIH, PHS, state law).
There are two places to find a link to the ICD Checklist:
- On the Education & Training page of the HSO website, under Educational Tools, or
- In the HawkIRB application, in the Help Message
 for the ‘Informed Consent’ section of the attachments page.
for the ‘Informed Consent’ section of the attachments page.
This checklist can also used by staff reviewers, compliance staff, IRB Chairs and IRB members when reviewing new applications, modifications and studies selected for monitoring. We strongly encourage use of the Informed Consent Document Checklist to ensure consistency between the HawkIRB application and the Informed Consent Document.
We hope that you will utilize this tool and find it helpful as you prepare informed consent documents for your research studies.
IRB Advisor, January 2018: Right to Try in Oncology: Gatekeepers or Mercenaries?
By Maegan Tyrrell
Bioethicists are concerned that with an increase in patient demand for investigational cancer drugs, there will be an increase in federal Right to Try requests. The Right to Try law, passed in 2018, is different than an expanded access approach in that it bypasses FDA and IRB review.
A possible outcome of an increase in Right to Try requests are ‘physician mercenaries,’ approving patients to pursue these drugs without knowing the patient. “The idea of physician mercenaries is problematic because it makes a farce of the gatekeeping role that Right to Try preserves for certifying physicians,” authors of a recent paper emphasized. The idea of mercenaries came up due to online pharmacies, where patients can get a prescription from a physician who doesn’t really know the patient. For Right to Try, patients need a physician to certify that they meet the eligibility criteria. “I think that physicians have a professional obligation to make sure patients are only pursuing Right to Try if it really is a reasonable pathway for them,” says Holly Fernandez Lynch, JD, MBE, Assistant Professor of Medical Ethics and Health Policy at the University of Pennsylvania.
IRB Oversight Role
Use of a non-FDA approved drug under the expanded access model requires FDA and IRB oversight, whereas Right to Try does not require either. Some researchers have questioned whether an IRB review and approval is necessary if the FDA is involved for the expanded access model. A recently published survey of IRB members found that 78% of respondents agreed that it is important for IRB review of single-patient expanded access requests. Lynch points out that IRBs review aspects of these cases that the FDA does not, such as the informed consent document and process. IRBs can also ensure that their institution has the right capabilities, resources, and support to oversee such cases. “You can debate whether their role is duplicative with expanded access, but with Right to Try I think the role of IRB review is important because you are losing FDA oversight,” Lynch says.
Other articles in the January issue include:
- Gene-Altered Twins Face Uncertain Future
- Money Matters: Payment to Research Participants ‘Haphazard’
- Need Researchers to Pay Attention? Try Experimenting With Engaging Content
- Study: Research Subjects Might Consent to Records Use, But Want to be Asked
- IRB Chairs Can Run Better Meetings by Following These Tips
- OHRP Gives IRBs a Break with Single IRB Review Exceptions
Current and Past Issues
IRB Advisor (a publication of Relias) is a monthly newsletter with articles about issues facing IRBs and individuals involved with human subjects research. The University of Iowa subscribes to this publication as a resource to UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff.
Current and past issues of IRB Advisor are posted in the “IRB ICON Course for Researchers” which is accessible to anyone with a UI HawkID and password. The portal to this ICON Course is on the Education and Training page of the Human Subjects Office web site. Due to our licensing agreement, we cannot post a link directly to this publication on the Human Subjects Office website.
In the News
First childhood flu helps explain why virus hits some people harder than others - Science Daily
Science can cure heartbreak in voles, but what about in humans? - WHYY
Vitamin D Deficiency during Pregnancy Connected to Elevated Risk of ADHD - University of Turku
Scrutinizing the effect of digital technology on mental health - Nature

