Index
From the Editor: The Informed Consent Issue, Part 2 (Plus the 10-year Anniversary of the HSRD Form)
Happy Holidays from the HSO!
FDA Issues Guidance for Enhancing Diversity in Clinical Trial Populations
Short Form Document and Process
External and Commercial IRB Q&A: How do study teams make/edit an Informed Consent Document for an external commercial IRB?
In the News
From the Editor: The Informed Consent Issue, Part 2 (Plus the 10-year Anniversary of the HSRD Form)
Kelly O’Berry, BS, CIP
The December 2020 issue of the IRB Connection Newsletter addresses additional topics related to informed consent that were not addressed in the November 2020 issue. This month we will cover:
- Informed Consent Documents (ICDs) for Industry Sponsored Studies using a Commercial IRB.
- Short form consent documents and process
This issue also includes an announcement about recent Food and Drug Administration (FDA) guidance regarding enhancing diversity in clinical trials.
Do I need IRB Approval?
This month, we are also celebrating the 10-year anniversary of the release of the Human Subjects Research Determination (HSRD) form. This very short form in the eResearch (HawkIRB) system is how researchers ask the most common question we are asked, which is: “I have an idea for a project. Do I need IRB approval for that?” You do not need to ask us this by email, over the phone, in the hallway… or at parties. The HSRD form collects all of the information an IRB Chair needs to provide the correct determination about whether the project meets the regulatory definition of human subjects research and therefore requires IRB approval.
When the Human Subjects Office and the Institutional Review Board began using this form in October 2010, we thought researchers would use it to ask if they needed IRB approval, when they were uncertain and needed an IRB determination before they began their project. However, we recently discovered that researchers also submit this form when they are pretty sure they do not need IRB approval, and they want a determination memo to document that.
The determination memo can be used to document that IRB approval was not needed if a researcher wants to publish or present the results of a quality assurance/quality improvement (QA/QI) project or a program evaluation, or some other project that does not meet the regulatory definition of human subjects research. This IRB determination memo is widely accepted by journal editors as indication that IRB approval was not required for the project. Usually within 2-4 business days of submitting an HSRD form, the IRB Chair will make a determination OR route the HSRD form back through HawkIRB workflow for further clarification.
You can learn more about the HSRD form on the Human Subjects Research Determinations page of the HSO website and in the February 2020 issue of the IRB Connection Newsletter.
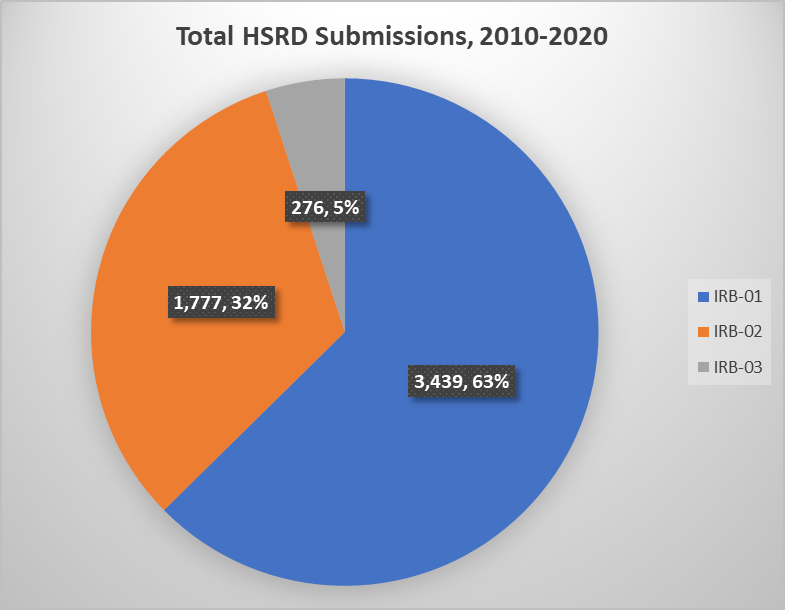
Between October 2010 and August 2020, the IRB received a total of 5,508 HSRD forms. IRB Chairs determined that 1183 (21%) were HSR and needed IRB approval and a whopping 4,325 (79%) did not need IRB approval.
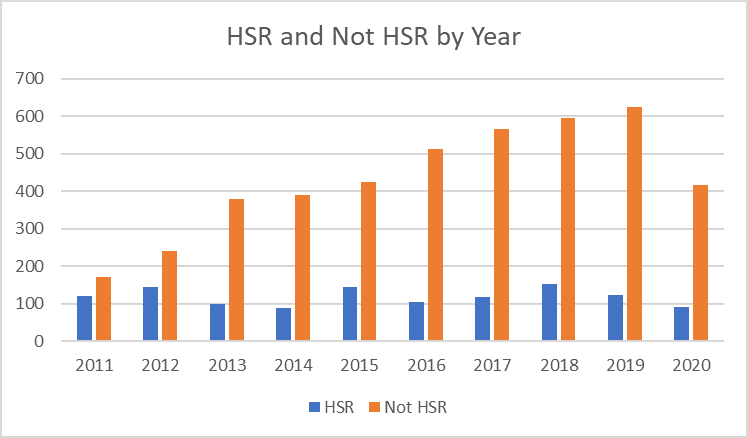
The number of HSRD form submissions grew steadily over the years, with the bulk of the submissions for IRB-01.
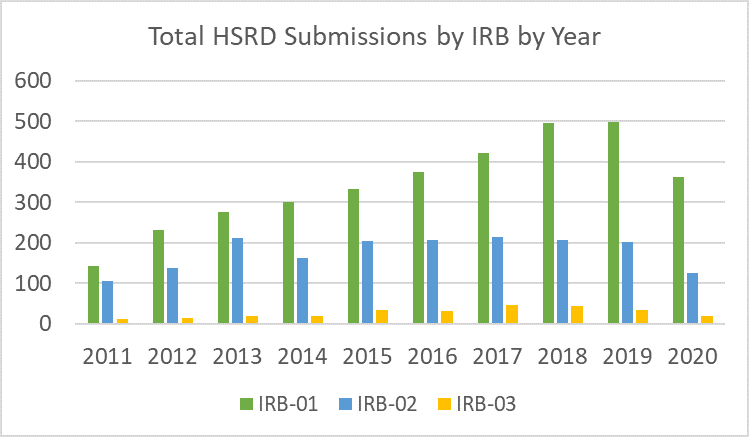
The majority of HSRD forms were submitted to IRB-01.
The HSRD form provides a valuable resource to help researchers know whether they need to prepare a HawkIRB New Project form and obtain IRB approval for a project. And it protects HSO staff reviewer and IRB time to review only the projects that meet the regulatory definition of human subjects research.
We publicly recognize the IRB Chairs who have reviewed the majority of HSRD forms and made these determinations over the past 10 years. A hearty THANK YOU! to IRB Chairs, Andy Bertolatus (IRB-01), Catherine Woodman (IRB-01 and IRB-03), John Wadsworth (IRB-02) and Janet Williams (IRB-02).
Contact the Human Subjects Office (irb@uiowa.edu or 319-335-6564) if you have questions or need assistance with completing an HSRD form.
Happy Holidays from the HSO!
Joanie Hoefer, BS, CIP
As the year 2020 ends, we in the Human Subjects Office would like to extend warmest wishes to all our research colleagues for a very happy and healthy holiday season.

We wish you and your loved ones all the best and we look forward to our continued collaborations in the brighter year ahead.
FDA Issues Guidance for Enhancing Diversity in Clinical Trial Populations
Joanie Hoefer, BS, CIP
In November 2020, the U. S. Food and Drug Administration (FDA) issued final guidance titled “Enhancing the Diversity of Clinical Trial Populations--Eligibility Criteria, Enrollment Practices, and Trial Designs,” that was first issued as a draft in 2019. This guidance outlines the agency’s current thinking about how to broaden eligibility criteria and increase enrollment of underrepresented populations in clinical trials.
This guidance considers both demographic (e.g., sex, race, ethnicity, age, etc.) and non-demographic characteristics (e.g., patients with organ dysfunction, comorbid conditions, disabilities, those at the extremes of the weight range, and populations with diseases or conditions with low prevalence) of study populations. The FDA provides recommendations in three areas so that sponsors may create study populations that more accurately represent the population that will take the drug if it is approved:
- Broadening eligibility criteria to increase diversity in enrollment. For example, relaxing more prohibitive exclusion criteria after Phase 2 trials are complete and including more children/adolescents, women and minorities to identify any differences in responses to a new medical product.
- Study design and conduct considerations for improving enrollment. For example, making it easier for people to participate by not requiring them to travel to a specific clinical trial site, use inclusive strategies to educate and engage the community and ensure there are trial sites in areas with higher minority populations.
- Broadening eligibility criteria and encouraging recruitment for clinical trials of investigational drugs intended to treat rare diseases or conditions. For example, engage patient advocacy groups, disease experts and patients themselves early in the design process for their input on the best way to design a potential study so that patients will want to participate and consider re-enrolling participants into later phases of the trial if safe and feasible to do so.
Short Form Document and Process
Kelly O’Berry, BS, CIP
One key requirement for research with human subjects is that researchers write consent document(s) and conduct the consent process in a language the subject can read and understand. Subjects should never be asked to sign a document that they cannot read. Researchers can use the Short Form Consent Document and consent process when they unexpectedly encounter a potential subject who does not speak English and the study does not have IRB-approved documents translated into the language the subject speaks.
This article will provide a general overview of the Short Form document and process and some important considerations for the Principal Investigator (PI) and research team before you enroll a subject using this process.
Short Form Consent Document and Process
The Short Form is an option for enrolling a non-English speaking subject when the research team did not plan for this in advance and does not have IRB approval for it. There are several requirements for the Short Form consent process:
- A thoughtful plan for ongoing communication with the non-English speaking subject
- Someone to interpret the information during the consent process based on the IRB-approved, English version of the Informed Consent Document
- A Short Form Consent Document translated into the language the subject speaks/reads
- A witness to the consent process (someone who is not a member of the research team)
- A plan for translating the full Informed Consent Document (if the study lasts longer than 60 days)
The University of Iowa IRB policy for the Short Form Consent Process is outlined in the UI IRB Standard Operating Procedures and Researcher Guide, Section II, Part 11.B.iii.a. A Short Form Consent Document Frequently Asked Questions (FAQ) and a Short Form Consent Document and Process Educational Tool (that provides step by step instructions and the English and translated Short Forms) are available on the Human Subjects Office (HSO) website. Studies using an IRB outside of the University of Iowa will likely have similar policies, however the researcher should review the respective IRB’s policies and procedures to ensure compliance.
Considerations Prior to Enrollment
Communication with the subject
The research team and the subject must be able to communicate with each other throughout the study. This requires identifying an interpreter for verbal communications and someone to translate written documents and study materials. These activities could be done by the same person or two different people or service providers.
The interpreter or translator could be a member of the research team. They may be a native speaker or have sufficient credentials to fully translate all verbal and written communications with the subject. The interpreter/translator could be a family member of the potential subject, a volunteer or someone hired by research team to provide this service.
When an interpreter or translator assists with verbal and/or written communications between the research team and the subject, they do not need to be named on the research team. They would not be considered “engaged in research” according to guidance from the Office of Human Research Protections. However, if the interpreter is actively involved in study activities, such as conducting the consent process, an interview or focus group with the subject, or answering questions about the study, they will need to be added to the research team.
When the study has more complicated procedures or technical descriptions, the interpreter or translator must have the knowledge base and language skills to accurately convey this information to the subject. For clinical trials or other complicated studies, this may require skills beyond just being a native speaker or a few years of language education. Additionally, when a study has multiple visits, an interpreter must be available for every study visit and for any communications to or from subjects between visits. All study materials provided to the subject throughout the study would need to be translated into the language they speak/read.
In the past, the UI Health Care (UIHC) Translation Service typically only provided translation for clinical care, not for research. Before enrolling a subject using the Short Form Consent Document and process, it is essential to identify an interpreter for the duration of the subject’s participation in the study.
IRB-Approval for Translated Informed Consent Document
If the study involves multiple visits over more than 60 days, the research team must provide subjects with an IRB-approved, translated Informed Consent Document. That means the research team must do all the following within 30 days of subject enrollment:
- Have someone translate the English versions of the IRB-approved document.
- Have a back translation completed on the translated Informed Consent Document.
- Submit a HawkIRB Modification form.
- Obtain IRB approval for the translated document.
- Mail this translated form to the enrolled subject.
The subject does not need to sign the translated consent document, but the research team should document in the research files that they provided the translated document to the subject. This can be done via a written note on the Short Form Consent Document or as a separate Note to File stored with the Short Form Consent Document. Check with the sponsor, if applicable, to determine the acceptable method for this documentation.
Translated Short Form
The Short Form Consent Document must be available in the language the subject speaks/reads. The English version is posted on the HSO website, accessible from the Short Form Consent Document FAQ. The document is currently translated into seven languages:
- Arabic
- Bosnian
- French
- Japanese
- Russian
- Spanish
- Swahili

External and Commercial IRB Q&A: How do study teams make/edit an Informed Consent Document for an external commercial IRB?
Jarrod Feld, BA
When a pharmaceutical company (or industry) sponsor submits an Informed Consent Document (ICD) template to a commercial IRB of record, individual sites then have the opportunity to revise the sponsor’s ICD to align with institution-specific requirements and language.
You should use the ICD template documents available in HawkIRB as a guide for the UI template language, especially regarding:

- Research-related injury
- Confidentiality protections
- HIPAA privacy statement
- Documentation of research participation in the University of Iowa Healthcare (UIHC) medical record (when applicable)
- The list in the signature section of who qualifies as a Legally Authorized Representative (LAR)
These sections must align with the UI template language and generally cannot be altered by the sponsor or the external/commercial IRB.
You may also use UI template language from other ICD template sections (when applicable):
- Entities who may have access to personal health information (e.g. ‘representatives’ is considered vague and not allowed)
- ClinicalTrials.gov
- Mandatory HIV/Hepatitis positive test reporting
- Commercial gain from future research on samples
- Whether tissue/specimen/data samples are optional or required
- Collection or use of social security number
- Whole genome/exome testing
- Pregnancy testing language for minors
- Use and sharing of Centers for Medicare & Medicaid Services (CMS), insurance, or other patient medical billing information
- Use of a home health care agency to conduct research activities
- Use of radiation as part of the research (governed by the Medical Radiation Protection Committee (MRPC))
In addition, review the sponsor template ICD for accuracy regarding study procedures, aims, risks, future use of data/specimens, etc. to make sure they align with the protocol and the contract for the UI site. There may also be UI site-specific language recommended by other Human Research Protection Program (HRPP) committees, such as UIHC Research Billing Compliance, MRPC, or the Pharmacy and Therapeutics Committee. The UI HRPP typically encourages the use of the term “subject” for research participant.
If the Informed Consent Document is modified during the review of a HawkIRB application for a commercial IRB, the PI will need to share the tracked changes version with the sponsor and/or Contract Research Organization (CRO) or Coordinating Center. They may suggest or insert additional edits. Some sponsors will include language not allowed in UI ICDs, especially with regards to accessing subjects’ personal health information (PHI). For example, language broadly referring to “other entities” having access to a subject’s PHI in the HIPAA section is not consistent with the regulations (45 CFR § 164.508(c )(1) - Uses and disclosures for which an authorization is required. The regulations require “the name or other specific identification of the person(s), or class of persons to whom the covered entity may make the disclosure.” Therefore, the UI HRPP requires language specifically describing a defined or demonstrated contractual relationship with the sponsor for any entities that may have access to PHI for the purposes of the research. Specific terms describing a contractual or fiduciary obligation to the sponsor could include:
- Affiliate
- Agent
- Contractor or subcontractor
- Employee
- Subsidiary
Terms that are seen as vague and not allowed include:
- Representative(s)
- Collaborator(s)
- Notified bodies
- Licensee
- Consultant
- Recipient
- Third party
- Others
This can be found in full in the ICD template document available in HawkIRB for studies involving a commercial IRB and\or industry sponsor.
It is also worth noting, in the majority of industry sponsored research, the sponsor is liable for all study costs, tests and procedures that are outside standard of care, study drugs/devices/equipment, as well as injuries that arise as a result of the properly performed study procedures. In accordance with CMS policy, if a sponsor indicates they will only pay for research costs beyond, or in addition to, what the subjects’ insurance will pay, this will not be allowed by the UI HRPP.
The consent language negotiation usually takes place during the review of the HawkIRB application. However, sometimes after the study team has submitted to the commercial IRB of record, the sponsor will make edits and the reviewers at WCG (WIRB) or Advarra (or other commercial IRB) will facilitate the consent language negotiations. It is important that UI study teams track the edits and additions sponsors may make after the project is submitted to the commercial IRB to ensure continuity with UI HRPP approved consent revisions. If the ICD is revised after the HawkIRB application is approved, submit a Modification form and use the Edit function to save the tracked changes version on top of the previous version.
When a Master Service Agreement is in place between the UI HRPP and a commercial IRB, it specifies the requirements for template language in the ICD. Therefore, it should not be a surprise to the commercial IRB that certain language must be included, or may not be included, in the final version of the ICD. However, it is the responsibility of the HSO review staff and the UI study team to ensure sponsor edits and additions are aligned with institutional, state, and UI-specific requirements.
In addition to the full ICD, the UI HRPP has applied the regulatory requirement of providing a summary of key information for all greater than minimal risk research, regardless of funding. Industry sponsors may include this at the beginning of the ICD or it could be a separate document. If the sponsor does not provide a template consent summary, use the General Consent Summary template in the drop-down menu for Consent/Assent Documents in the HawkIRB system.
Keep in mind that every study is different. Every sponsor has different rules and procedures. Additionally, the various commercial IRBs have their own processing and communication guidelines. We strongly recommend that study teams reach out to the Human Subjects Office staff and review the information on the HSO website for further guidance for multi-site studies utilizing a single/central IRB of record. Our emails for these studies are UIWIRB@iowa.edu (for WCG/WIRB and Advarra studies) or uirb-external@uiowa.edu for studies utilizing another IRB of record.
In the News

- Ethics of Placebo Controls in Coronavirus Vaccine Trials, The Hastings Center
- Placebo-Controlled Trials of Covid-19 Vaccines – Why We Still Need Them, New England Journal of Medicine (NEJM)
- Emergency Use Authorization for Vaccines Explained, Food and Drug Administration (FDA)
- Unequal Representation: Race, Sex and Trust in Medicine – COVID-19 and Beyond, Bill of Health, Harvard Law Petrie Flom Center
- Coronaviruses hijack lysosomes to exit cells, NIH Research Matters

