Enrolling Children in Research: They’re not just small adults!
by Kelly O'Berry

There are compelling reasons for including children in research but there also may be heightened concerns about the safety and welfare of younger research participants. This article will cover the regulatory requirements for conducting research with subjects who are less than 18 years of age.
Whether a study is conducted in a biomedical or social behavioral field, most research involves at least some level of risk to participants. Researchers are required to make every effort to minimize risks to subjects of all ages. Although children may face risks from participating in research, they face additional risks when physicians prescribe medications that were only tested in adults. According to the FDA, only about 20% of drugs approved by the FDA are labeled for pediatric use and physicians regularly prescribe medications to children “off label” not knowing the effects of the medication or the appropriate dose for children. Enrolling children in research is the only way to further scientific knowledge about the effects of medications and about diseases and conditions that affect children.
Credit: Zachary Scott, New York Times (1981)
Federal Regulations
Subpart A (the Common Rule) of the Department of Health and Human Service regulations for human subjects research and Section 21 CFR 50.3 of the Food and Drug Administration (FDA) regulations introduce the concept of a Legally authorized Representative to give permission for another person to participate in research. Subpart D of the HHS regulations specifies that for children, the only people who can serve in this capacity are the child’s parent or a legal guardian (appointed by the court if parental rights are terminated).
The Common Rule (45 CFR 46.111) and the FDA regulations (21 CFR 56.111) both specify eight criteria that all studies must meet to be approved by the IRB. Several of these criteria apply to research studies that enroll children:
- Minimizing risks to subjects – Researchers must use procedures consistent with sound research design. And risks may be minimized by using procedures that are already performed on subjects for diagnostic or treatment purposes.
- Risk/benefit evaluation – The risks to subjects must be reasonable in relation to anticipated benefit, if any, to subjects, and the importance of the knowledge that may be gained from the study.
- Informed consent will be sought and documented – The research team will conduct the consent process and document subject agreement to participate in the research, unless the IRB waives these requirements.
- Additional safeguards for vulnerable subjects – A researcher may need to implement additional protections for subjects who are vulnerable to coercion or undue influence; who may not be able to make a decision about participation that is in their own best interest.
In 45 CFR 46.110, there is a link to a list posted in the Federal Register with categories of research that qualify for IRB expedited review procedures. This list includes limits on the amount of blood researchers can collect from subjects depending on their age, weight and the health of the subject.
The HHS regulations include three subparts that address research with vulnerable subject populations. Subpart D outlines additional protections for children involved as research subjects. That includes restrictions on the type of research that can be approved by the IRB based on the level of risk and the potential benefits to the minor subjects. This section includes the following definitions”
- Child - a person who has not yet attained the legal age for consenting to research activities
- Parent - the child’s biological or adoptive parent.
- Guardian - anyone authorized under applicable state or local law to consent on behalf of the child for medical care.
Parent/Guardian Permission
Usually, adult subjects can give informed consent to participate in a research study. A child cannot give legally effective informed consent for their own participation in research. However, the regulations allow a parent or legal guardian to grant permission for a child to enroll in a research study. A child may live with someone other than their parent (a grandparent or aunt/uncle), but unless parental rights have been terminated and the court has appointed a guardian, the research team must obtain permission from the biological or adoptive parent. For research involving children as subjects, researchers must be vigilant about obtaining and documenting permission from someone who is legally authorized to give it.
Wards of the State
There are additional restrictions if minor subjects are Wards of the State. This includes children who have been removed from parent custody by Child Protective Services and placed in foster care or a juvenile detention facility. In Section VI.11 of the HawkIRB application, UI researchers indicate if they plan to enroll foster children or Wards of the Court. If so, they must describe the subjects AND explain how they will obtain consent from the parent or legal guardian. If the researcher has not provided this information to the IRB, the study is not approved to enroll children who are in foster care or Wards of the Court. The researcher may submit a HawkIRB Modification form to provide this information and request IRB approval to enroll these subjects.
Enrolling children in research requires some additional considerations and planning that are not necessary for research with adult subjects. Researchers must consider the needs and abilities of both the parents/guardians and the children themselves when planning the recruitment and consent process and the study procedures. Do not expect children to be like “small adults” as research participants.
Best Practices for Documentation of Informed Consent
by Patricia Katopol, PhD
For most human subjects research, Federal regulations require that volunteers provide informed consent before participating. Whether from the subject or the subject’s legally authorized representative (LAR), documenting informed consent can present challenges for investigators, study coordinators, and other team members. This article describes selected best practices that may improve the documentation of consent for all research subjects.
Some documentation issues with Informed Consent Documents (ICD) are easy to overlook. For example, a consent returned by mail may have only the signature page. If that happens, print out the missing pages of the consent and attached them to the signature page. Make sure the approval stamp appears on all pages, not just the first page, and check that the subject or LAR signs the correct version of the ICD.
Valid Consent Documents
One way to ensure that the correct ICD form is used is to download the consent from the attachments tab in HawkIRB, not from a downloaded consent saved to your computer. Only the current, IRB-approved, consent should be used. It is an important part of good documentation practice to verify that the consent is signed within the date range in the grey box at the top right of each page. The IRB approval stamp includes the date the document was approved and the date IRB approval expires. These dates are updated every time a new version of the consent document is approved; either via a HawkIRB Modification form to change the consent document or when a Continuing Review form is approved. Best practices will include methods for keeping team members aware of these changes. If a consent is not current, it is not valid. If invalid consents come to the attention of the Board, it may disallow the use of data collected from those subjects.
Documentation of Consent By Mail
Mailed consents have unique documentation issues because usually there is no member of the research team with the subject to make sure printed names and signatures are in the right place or that subjects correctly indicate their choice on optional agreements. If using a mailed consent process, the research team should do two things:
- Write clear instructions for subjects to sign and return the ICD, and
- Establish a plan to correct errors in the signatures or in marking optional agreements.
The correction plan should be described in HawkIRB Section VII.D.29 and VII.D.30. There is a template correction letter in the dropdown menu of the Miscellaneous Attachment category on the HawkIRB Attachment page. Use it to create a template letter to send to subjects to correct errors in ICDs that are returned by mail. Since they are used to communicate with research subjects, the instructions and correction letter must be approved by the IRB.
Study Coordinator Panel Discussion
Project coordinators are often directly involved with obtaining consent, managing signed consents, and overseeing team members who obtain consent. On March 28, 2017, the Education and Outreach Group of the Human Subjects Office presented a panel discussion, Informed Consent: Best Practices from Study Coordinators. Our panelists were Cathy Fairfield, Department of Emergency Medicine; Debra O’Connell Moore, Institute for Clinical and Translational Science Clinical Research Unit; and Chris Sinkey, Ophthalmology and Visual Science.
These seasoned coordinators offered useful insights about how they approach documenting consent:
- Pay Attention When the Subject Signs - Avoid subject errors in signing and dating by paying attention while the subject is signing the ICD. Make sure they respond to optional agreements, sign on the correct line, provide the correct date for their signature, etc.
- Be Respectful of Subjects - When consenting during medical emergencies, be mindful of prospective subjects’ situation. A patient in the Emergency Department or who has just received a serious medical diagnosis may not be open to hearing about a research study. The research team should be respectful of the availability and any special needs of these potential subjects. Recognize that the subject is a person who deserves your attention and respect.
- Thoroughly Train Everyone Involved in Conducting the Consent Process – Team members who are new to consenting and /or working with people who are experiencing illness or uncertainty about their health will benefit from training by senior coordinators or other experienced team members on consenting in stressful environments.
- Consent Documents - Informed Consent Documents should be written in plain language at the eighth grade reading level. It may be useful for the PI or coordinator to give a draft consent to another staff person – or even a child if writing an assent – to see where comprehension problems may arise and to determine if the intended population will understand the study and their role in it. Team members who obtain consent should be aware of cultural differences that may affect subject behavior during the consent process. For example, if the subject’s culture dictates that it is impolite to ask questions, the person obtaining consent should be proactive in eliciting information from the subject to ascertain that s/he understands what is in the ICD.
- Permission from Parent/Guardian or Legally Authorized Representative (LAR) - When working with an LAR, make sure the person actually has the authority to give permission for a child or cognitively impaired adult to participate in the study. See LAR Legally Authorized Representative for more information about LARs in Iowa.
Panelists and attendees agreed that the most important takeaway from the presentation was the importance of creating trust with the subject. With a trusting relationship, the subject may share important or sensitive information, and it can help with their retention in the study.
The proper documentation of consent is an important part of human subjects research. It can present human, technological, and administrative challenges; however, panelists and audience members agreed that adopting best practices for documentation could greatly reduce them.
External/Commercial IRB Q&A Info Spot: Changes to the HawkIRB Modification Submission Requirements for Studies with a Commercial IRB of Record
by Kathy Beck

Q: Do I need to submit a HawkIRB modification for studies with a Commercial IRB of Record?
A: Yes. The UI investigator is responsible for submitting a HawkIRB Modification form when study revisions require local review. The UI Human Research Protection Program (HRPP) recently revised the requirements for local review. The Human Subjects Office (HSO) will no longer require submission of HawkIRB Modification forms for every modification reviewed and approved by a commercial IRB of Record. The UI HRPP only needs to review the following categories of modifications:
- Changes or additions to:
- Consent documents: the consent, assent, other consent documents
- Protocols, including clarifications, letters, and summary documents
- Investigational Brochures
- Device-related Investigational New Drug (IND) or Investigational Device Exemption (IDE) documents
- Research team member changes
- Mass email or noon news requests
- Correction notices issued by the IRB of Record. For example, if the external IRB issues a revised/corrected approval notice, it will include the type of correction. Corrections can include administrative changes and additional documents reviewed.
- Changes to the current external IRB application content or a “yes” response to a question in a HRPP Committee checklist in Section XIV.
- Section XIV contains checklists for the Human Research Protection Program (HRPP) committee(s). Depending on the study, applicable checklists will be required:
- Pharmacy & Therapeutics (P&T) Committee, Medical Radiation Protection Committee (MRPC), the Joint Office for Compliance (JOC), Holden Comprehensive Cancer Center Protocol Review (PRMC), and Nursing Research Committee (NRC)
- Section XIV contains checklists for the Human Research Protection Program (HRPP) committee(s). Depending on the study, applicable checklists will be required:
- Changes related to policies and procedures outlined in the UI Operations Manual. The HawkIRB application will need to be updated and, if applicable, HRPP Committee review(s) may be required.
All other types of modifications should be submitted to the commercial IRB of Record but do not need to be submitted in a HawkIRB modification form. The HRPP review process remains the same for all changes that meet the requirements for submission in HawkIRB. Additional information is available on our website.
Q: Do I need to submit a HawkIRB modification when the sponsor submits a modification on behalf of the UI investigator?
A: Yes. The UI investigator is responsible for submitting a HawkIRB Modification form when study revisions require local review, regardless of who submits the modification to the commercial IRB of Record.
Q: Why did the requirements change for submitting modifications?
A: Based on feedback from our investigators and HRPP Committees, the Human Subjects Office (HSO) revised the requirements for researchers to submit modifications for approved studies under the purview of an external IRB of Record. The effective date was April 25, 2017. We anticipate these changes will provide a significant reduction in effort by research staff and still address all HRPP review requirements of human subjects research. The modifications that do require tracking are associated with either an institutional policy or a human research protection program review requirement.
If you have additional questions about submitting modifications for studies under the purview of a commercial IRB of Record, please contact Kathy Beck (335-7297, uiwirb@uiowa.edu).
International Research: IRB Approval in Any Language
by Brent Collinsworth
University of Iowa faculty, staff, and students always need UI IRB approval for all human subjects research, even when it is conducted outside the United States. If you are planning to conduct human subjects research in another country, you will need some additional approvals prior to submitting your HawkIRB New Project Application. This includes getting in-country ethics review committee approval, obtaining a local context review, and/or getting permission to conduct research in a specific location and obtaining a letter of agreement.
In-Country Ethics Committee Approval
First, you will need to check if your project needs approval from an in-country ethics committee. It is the researcher’s responsibility to identify and ensure compliance with all regulations, committees, laws, and guidance concerning human subjects research in their target country. The Office for Human Research Protections issues an International Compilation of Human Research Standards every year that outlines the key organizations, legislation, regulations, and guidelines for over 100 countries and several international organizations, such as the World Health Organization. Anyone planning to do research abroad will need to review the Compilation and obtain all necessary approvals before IRB submission.
Next, you may also need approval from a local or institutional ethics committee. For instance, if you are conducting research at a foreign university, you may need approval from that university’s ethics board. This will not be specified in the International Compilation, so you will need to ask the institution where you are conducting research if an ethics committee review is required.
Finally, note that having ethics approval in that country/site does not override getting ethics approval from the University of Iowa IRB. The UI IRB has ultimate responsibility for human subjects research conducted by UI faculty, staff, and students, so local IRB approval is required prior to conducting any research activity, regardless of where it is conducted. You must obtain in-country ethics approval before submitting the study to the UI IRB. If required, attach documentation of local ethics board approval to your HawkIRB New Project Application.
Local Context Review
If you do not need to get formal in-country ethics approval for your project, you will have to provide what is called a “local context review.” This step is designed to verify that your research in that country is both ethical and culturally appropriate. You will need to identify a local context reviewer who has expertise in conducting human subjects research in that country, and they will need to provide their qualifications to the IRB as part of the local context review. In order to avoid bias, you cannot complete your own local context review.
The local context review will address:
- What risks, if any, subjects face from participation in the study
- Are the consent materials and overall consent process appropriate for the subject population?
- If children or minors are involved, is the consent/assent process appropriate?
- Are there any in-country regulations related to human subjects research?
You can generate the template local context review document from the drop down menu in the Miscellaneous Attachments section of the Attachments page of your New Project Application. Once it is complete, add it as a “Miscellaneous Attachment.”
Permissions/Letters of Agreement
If you indicate in Section VII.A.1 that you are conducting your research off campus or outside the United States, the HawkIRB application will open a category for you to attach a Letter of Agreement documenting permission to conduct the research in that location. If you are planning to conduct research in a specific agency, organization or location, or with specific resources (such as staff) from that agency/organization, to the Letter of Agreement shows the IRB that you have permission to do so. The Letter of Agreement indicates that someone at the agency or organization gave permission for you to conduct your research there. You can find instructions and a template letter of agreement in the drop-down menu of the Miscellaneous Attachments section of the HawkIRB Attachments page. This letter needs to establish three things: the person writing it
- has the authority to give permission,
- understands the research (either giving a short summary of the research procedures or acknowledging the researcher’s summary of events), and
- grants permission for the research to occur there.
There are certain locations for research activity that you may not be able to obtain permission in advance, such as a public park or the subject’s home. If you are conducting your study in a location where it is not possible to obtain a Letter of Agreement, you can simply leave the Letter of Agreement section of the Attachments page empty. You will get an error message when you submit the application, but the HawkIRB system will let you submit the form without an attachment in that category.
In summation, when doing international research, you need additional approvals before you submit a New Project Application to the IRB. Make sure you start this process as far in advance as possible. You cannot submit your New Project Application until all of the other documentation and approvals are in place. Be sure to leave yourself enough time to obtain UI IRB approval before you leave the U.S. to conduct your research.
Do Quality Assurance/Quality Improvement Projects Need IRB Approval?
by Kelly O'Berry
Quality Assurance/Quality Improvement (QA/QI) projects typically do not fit the definition of human subjects research, because they are not intended to “contribute to generalizable knowledge.” However, UI researchers who do QA/QI projects are strongly encouraged to ask if they need IRB approval prior to initiating the project. This article includes a description of QA/QI projects and provides guidance on asking whether IRB approval is required.
University of Iowa researchers use the Human Subjects Research Determination (HSRD) form in the HawkIRB system to ask if a specific project meets the regulatory definition of human subjects research. Within two or three business days, an IRB Chair typically makes a determination or returns the form through HawkIRB workflow with questions for the researcher to address.
A definition of Quality Assurance/Quality Improvement
QA/QI projects are systematic, data-guided activities that seek to evaluate or identify ways to improve quality, outcomes, system performance or any other kind of improvement to an existing service. In QA/QI projects, providers and/or participants evaluate their daily experiences to identify ways to improve care or services and implement changes. The project may involve collecting data on the effects of those changes and assessing the results.
Differences Between QA/QI and Research Projects
| Points to Consider | Research | QA/QI |
|---|---|---|
| Purpose | To test a hypothesis or establish clinical practice standards where none are currently accepted | To assess or promptly improve a process, program or system; OR improve performance as judged by accepted/established standards |
| Starting Point | To answer a question or test a hypothesis | To improve performance or quality of a service |
| Benefits | Designed to contribute to generalizable knowledge; may or may not benefit subjects | Designed to promptly benefit a process, program or system; may or may not benefit patients or clients |
| Risks/Burdens | May place subjects at risk and risks must be disclosed to subjects | By design, projects do not increase patient/client risk, with exception of possible privacy/confidentiality concerns |
| Data Collection | Systematic data collection through intervention or interaction | Systematic data collection through interaction |
| End Point | Answer/address a research question | Promptly improve a program or process or system |
| Testing/Analysis | Statistically prove or disprove a hypothesis | Compare a program or process or system to an established set of standards |
*Adapted from QA/QI guidance from the Stanford University Human Research Protection Program
The HSRD Form
UI researchers submit an HSRD form in HawkIRB when they are not sure whether IRB approval is required. [NOTE: Some researchers submit HSRD forms even if they are pretty sure they don’t need IRB approval, just to obtain formal documentation of that determination from an IRB Chair.] Documentation about whether IRB approval is necessary may be requested by a sponsor or funding agency and can be provided to scientific journals when submitting a manuscript for publication. Yes, it is possible to publish a QA/QI project even if it did not need IRB approval.
The HSRD form has a series of questions to identify and describe plans for QA/QI projects. One of the questions is HSRD I.7:
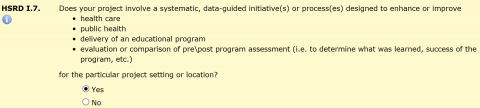
This question only opens if you indicate in HSRD I.5 that the study is not intended to contribute to generalizable knowledge. If you answer “Yes” to HSRD I.7, a series of five additional “Yes/No” questions collect the following information:
HSRD I.8 - Purpose of the Project
If the researcher has a hypothesis or a research question and intends for the results to further scientific knowledge in a particular field of study, the project will probably meet the definition of human subjects research and IRB approval will be required. However, the purpose of a typical QA/QI project is to improve the process or delivery of a program or service. The results may be shared with the agency or organization that oversees the program or service. If the proposed project is intended to improve a process or delivery of a program or service, the IRB Chair will look for two types of documentation.
1. Specific literature or other background reference included in section I.4.a of the HSRD Form. The IRB would expect this literature or reference would provide an indication that the process improvement was successfully implemented elsewhere and the intention is to implement this documented standard in a new location.
OR
2. There is a business purpose to implement the proposed activity in place of a current practice. The IRB would expect to see relevant leadership at the location of the proposed activities. Generally, documentation of this would come from a Privacy Officer, CEO, or President and be attached in on the Attachment page of the HSRD form.
HSRD I.9 - Project Staff
A Principal Investigator and members of a research team conduct research activities according to the study protocol. The HawkIRB application for this type of study will describe the study design and procedures so the IRB can determine whether the projects meets the criteria for IRB approval (45 CFR 46.111). A QA/QI project is often conducted by staff who provide services, care or education or are responsible for the quality of the services provided in that setting.
Please note: The University of Iowa IRB does not have the authority to provide a QA\QI or HSRD determination for a non UI entity affiliated with another IRB of record. In these cases, the IRB of record must be consulted for an official determination.
HSRD I.10 - Recruitment
Research studies have specific eligibility criteria for the subject population and a plan for how potential subjects will be identified and invited to participate. A typical QA/QI project will involve collecting information from staff and/or patients, clients or students in setting where the project will take place.
HSRD I.11 - Informed Consent
The ethical foundation of human subjects research is asking if an individual wants to participate in a research study and giving them enough information to make an informed decision about whether or not to enroll. In a QA/QI projects, typically no additional permission or consent is required beyond their agreement to engage in the program services, education curricula or clinical care setting.
HSRD I.12 - Risks
In human subjects research projects, researchers must minimize the possible risks to subjects from participating in the study. When deciding if a project can be QA/QI rather than human subjects research, an IRB Chair will evaluate whether there are any risks to participants beyond those they face in the services, care or education they are already receiving.
Important Additional Information
Projects that meet the definition of QA\QI are still required to address HIPAA if accessing protected health information. The owner of the data (i.e. generally the designated Privacy Officer of the facility) must explicitly grant authorization to access any PHI. Under the HIPAA regulations (45CFR46 164.506) regarding “Uses and disclosures to carry out treatment, payment or health care operations,” the entity must document and agree the QA\QI project is consistent with the definition of “Health Care Operations” as defined under 45CFR46 164.501. In this instance, IRB-01 would not have the authority to grant these authorization under HIPAA. IRB-01 only has the authority to grant waivers of HIPAA authorization when the project meets the regulatory definition of human subjects research. Contact the Human Subjects Office to request a template letter to be signed by the HIPAA Privacy Officer of the agency or organization.
There is more information about the HSRD form on the Human Subjects Office (HSO) web site. Researchers may come to IRB Office Hours or contact the HSO by phone or e-mail for additional assistance with completing an HSRD form (Ph: 319-335-6564 or E-mail: irb@uiowa.edu).
Herky Hint: Getting Approval to Access Medical Records for Recruitment Purposes
by Therese Barenz, BSN, CIP

A key component of the Privacy Rule is that it applies only to PHI held or maintained by a covered entity or the covered components of a hybrid entity. A covered entity (or covered components of hybrid entity) may use or disclose PHI for treatment, payment, or operations – but not for research activities. The Privacy Rule does not apply to research. However, if a research investigator is part of the covered entity, HIPAA rules apply to accessing PHI for the research study. The UI is a hybrid entity because it is a single legal body with both covered* and non-covered components.
What does all this mean?
Investigators may use and disclose PHI held by a covered or hybrid entity for research purposes if they get a subject’s written permission in the form of an authorization. For studies at the UI that involve obtaining PHI from UI\UIHC hybrid covered entities as part of study procedures, the HIPAA authorization is incorporated into the ‘WILL MY HEALTH INFORMATION BE USED DURING THIS STUDY?’ section of the Informed Consent Document (ICD) that the IRB reviews and approves. Researchers obtain permission to access this PHI when individual’s sign the consent document.
When a researcher plans to access PHI for recruitment purposes before consent, s/he won’t have the subject’s authorization. The good news is that the Privacy Rule allows IRBs, designated as Privacy Boards, to review requests to alter part or all of the Authorization requirements to use or disclosure PHI for research studies and provide documentation of the waiver. IRB-01 serves as the Privacy Board for the UI\UIHC hybrid covered entities only; it cannot provide approval for non-UI entities. If UI researchers need to access PHI at a non-UI/UIHC hybrid-covered entity, they must obtain approval from the respective Privacy Board and provide the UI IRB with the documentation. The UI provides a sample letter for PIs to use to document compliance with the HIPAA Privacy Rule when working with non-UI covered entities.
The official term for the approval to use PHI prior to consent is a ‘partial waiver of HIPAA authorization’ for recruitment. Researchers request and provide justification for this partial waiver in the HawkIRB application.
How Do I Request the Partial Waiver of HIPAA Authorization for Recruitment Purposes?
In Section VII.D.1 of the HawkIRB application, select the following recruitment method:
“Use of any information available to the researchers or their colleagues because this person is a patient OR use of any information considered to be Protected Health Information (PHI) OR review of patient/clinic records, Describe source of records”.
Sections VII.D.2-7 will open to provide justification for the partial waiver of HIPAA authorization. Your responses in these sections are significant because they demonstate to the IRB that:
- You could not conduct the research without access to this PHI AND
- You have an adequate plan to protect the information and assure that it won’t be used/disclosed outside the research team.
A central principle of the Privacy Rule is that users will limit the amount of PHI they access and disclose to the minimal amount necessary to accomplish the intended purpose. Section VII.D.2 is asking researchers to think about ‘What is the minimum amount of information we need to determine if someone would qualify for our study?’ More specifically, “What individual items do we need to review in the medical record to decide whether to invite the person to participate in the study?” This list should be limited to, and mirror, the information in the inclusion/exclusion criteria provided in Section VI.13 (e.g., diagnosis, age, medications, pathology/radiology results, lab results.) It should not list the medical information the research team will collect after consent. The collection of that information will be described in the study procedures section in VII.E.6.
Section VII.D.3 asks why the researcher could not recruit subjects without access to and use of PHI. “Practicably” means that it would not be possible. The IRB cannot grant the waiver if it would just be inconvenient for the researcher to ask the patient for permission to access their medical record OR if the researcher thinks patients would not approve this access.
In this section, you need to explain why it is not possible to identify subjects for recruitment without access to the PHI listed in VII.D.2. Appropriate responses would be:
- The eligibility criteria include …and list the specific diagnosis, specific lab – positive test or genetic result. We would not know whom to recruit without this information.
- It is not practicable to approach all the clinic patients when only a limited number would be eligible based on the inclusion criteria (e.g., diagnosis of specific type of cancer).
Section VII.D.4 asks why it would not be possible to get permission from individuals to look in their medical record. Appropriate responses would be:
- It is not practicable to obtain consent from all patients seen in the clinic, when only a small percentage will be eligible based on the specific eligibility criteria (e.g., diagnosis of xyz or a rare condition in medical history) listed in VII.D.2.
- (For research team members who are clinicians) We have access to the PHI because the potential subjects are my patients. [Note: This may not be true for all research team members who will access PHI.]
Section VII.D.5 asks to describe plans to protect the identifiers from improper use or disclosure. Since you are looking in the medical record to recruit subjects, the plans should describe confidentiality measures used to protect the limited data (listed in VII.D.2) that the research team obtains for recruitment purposes, BEFORE consent is obtained from subjects. The method will depend on the type of data you record – paper or electronic. If no information is printed or recorded from the medical record, then state, “No information is printed or recorded”.
The description should NOT describe:
- The use of a study ID code as this is normally assigned after enrollment in the study.
- The storage of research data that is obtained after consent (e.g., plans for storage of consent documents and research records).
Researchers are not authorized to keep PHI on individuals who do not enroll in the study. Usually the research team must remove the identifying information as soon as the subject refuses to participate. With adequate justification, the IRB may approve keeping the information through the recruitment/enrollment period in order to avoid approaching individuals who either declined to participate or were determined to be ineligible to enroll in the study.
If the response in Section VII.D.5 was, that research team does not print or record PHI from the medical record prior to consent; the response to VII.D.6 should state that there are no identifiers to remove or destroy.
IRB Approval of a Partial Waiver of HIPAA Authorization
Once the HawkIRB application is submitted, the IRB will review the responses to Sections VII.D.2-7 to determine if the use and disclosure of the PHI described involves no more than minimal risk to individuals’ privacy based on these criteria. Per the HIPAA regulations (45 CFR 164.512(i)(1)(i)), if the IRB determines that a study qualifies for the waiver, it makes a regulatory determination to waive the requirement to obtain an individual’s authorization (signature). This allows the research team access to a limited (partial) amount of PHI related to the study eligibility criteria, to identify potential subjects for the study. Upon approval, the IRB will attach a Partial HIPAA Waiver memo in the HawkIRB form Approval Tab and, when applicable, the IRB meeting minutes will document this IRB determination.
*Health care components of the UI hybrid entity: UI Health Care, College of Dentistry, College of Nursing, Wendell Johnson Speech and Hearing, Seashore Hall Psychology Training Clinic, UI Belin Blank Center, UI Wellness Office and Department of Athletics.
IRB Advisor June 2017 Digest
This month, we will highlight multiple articles from the June IRB Advisor newsletter. ![]()
Using Prisoners as Research Subjects Raises Ethical Concerns
Often, researchers and ethics committees assume that prisoners face a high risk of being coerced into participation in research. However, the results of a survey conducted by Dr. Paul P. Christopher, assistant professor of psychiatry and human behavior at Brown University, might change that assumption. Out of the 55 prisoners that agreed to be interviewed, he found that none felt coerced to participate in a study they volunteered for. In fact, around thirty percent of prisoners felt that they were pressured not to participate in a study. For a variety of reasons (including publicizing of data, loss of confidentiality, and potential for mistreatment), these prisoners were encouraged not to participate in research by other prisoners and correctional staff.
Future World Without Paper Consent Could Be Here Sooner Than Imagined
In the world of ever-present internet connectivity, social media, and touchscreen computers, consent documents printed on paper may one day go the way of the dinosaur. However, some experts are predicting that future to be sooner than expected. Some predict that, within seven years, most clinical trials will use some form of digital consent process. This process has some clear advantages (the ability to consent subjects without using research team members, for one), but will need to avoid some major pitfalls before widespread adoption is a reality. These pitfalls include the large upfront cost of designing electronic platforms and compliance with federal research regulations and national data privacy laws, which are discussed in more detail another article, Electronic Consent Has a Few Obstacles and Drawbacks.
Self-certification Tool Formalizes Process to Decide Between QI and Research
IRB officials at the University of Wisconsin-Madison realized they needed to solve a major problem. In 2010 they came to the conclusion that they were receiving many requests to review studies that were clearly quality improvement research, where IRB approval is not required. In 2013, they began using a tool that would allow faculty, staff and students to self-certify that their research did not need IRB approval. In the first year of using this tool, total submissions dropped 30%. Total submissions dropped another 30% in the subsequent year. A sample of the tool is available in the article, Questions From UW Madison’s QI Program Evaluation Tool.
Read the June issue of IRB Advisor, which also includes an article about the low rates of researchers reporting research misconduct when conducting systematic reviews.
What is IRB Advisor?
IRB Advisor is a monthly newsletter that contains articles about regulatory issues, informed consent, current events in human subjects protections as well as articles about IRB administrative and management issues. The UI IRB subscribes to this publication as a resource for UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff. Each month the IRB Connection Newsletter features an article from the current issue of IRB Advisor.
Current and Past Issues
There is a link to current and past issues of IRB Advisor on the Education and Training page of the Human Subjects Office web site. This link provides automatic access to the newsletter from all computers with a University of Iowa IP address.
UI faculty, staff and students and VA researchers who want to access IRB Advisor from a personal computer may contact the Human Subjects Office to request the username and password. Contact us by e-mail (irb-outreach@uiowa.edu) or call us at 319-335-6564.
Continuing Education Credits
Individual newsletter subscribers can receive 1.5 AMA PRA Category 1 Credits™ or 1.5 nursing contact hours for reading an issue and completing an online test. However, since this is an institution-level subscription, UI researchers must purchase access to an individual issue for $40, or purchase a full subscription, to receive CME/CE credits. Visit the AHC Media web site for information about subscription options.

