From the Director
In the event you missed our Fall 2016 three-part educational series on IRB Reliance models & processes, you are in luck! This month’s IRB Connection has you covered. In this newsletter, expect to see information on the following items:
- Why the use of a single IRB is all the rage in the article titled “Use of a Single IRB of Record”
- What you need to know about single IRB of record scenarios in the article titled “What’s All the Fuss? External IRBs and the Single IRB of Record Models”
- Who and what do you need to know about commercial IRBs in the article titled “What Does It Mean to Rely on a Commercial IRB?”
- What you need do as a Principal Investigator of a multi-site research project in the article titled “PI Responsibilities when utilizing a Single IRB”
- How to complete the IRB application in HawkIRB regardless of whether you serve as the lead site with the UI as the IRB of record OR you are relying on another IRB in this month’s Herky Hints.
I want to thank you all for subscribing to our monthly IRB Connection. As always, please contact our office if you have any questions in using or setting up an IRB Reliance model.
Michele Countryman, Human Subjects Office Director
Use of a Single IRB of record

By Laura Dallas and Martha Hedberg
Since federal regulations for protection of human subjects were first enacted in the 1970’s, IRB review has primarily been conducted separately by each institution participating in the research. This model for individual IRB review and oversight of the same study conducted at multiple sites is expanding with a new approach. This new method enables one IRB to extend review and oversight to multiple research sites.
This relationship is often referred to by various names, such as “Single IRB Model,” “Central IRB,” or “Shared IRB” review. The use of a single IRB (sIRB) for review of research at multiple institutions is intended to increase efficiency. Proponents argue that every IRB refers to the same set of federal regulations for the review of research, so it makes sense to have just one IRB review and oversee a project. Reviewing research from multiple institutions by a single IRB (sIRB) model does take some of the workload off those relying IRBs; however, the sIRB responsible for the review of multiple sites will experience a more complex review.
On June 21, 2016, the National Institutes of Health (NIH) issued the Final NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research, which establishes the expectations for use of a single IRB (sIRB) of record for:
- Non-exempt human subjects research protocols
- NIH funded research
- Research proposing to carry out research related activities at more than one research site in the United States
- Multi-site studies where each site will conduct the same protocol
The objective is to enhance and streamline the IRB review process in the context of multi-site research so that research can proceed as effectively and expeditiously as possible. Eliminating duplicative IRB review is expected to reduce unnecessary administrative burdens and systemic inefficiencies without diminishing human subjects protections. The shift in workload away from conducting redundant reviews is also expected to allow IRBs to concentrate more time and attention on the review of single site protocols, thereby enhancing research oversight.
The sIRB review must comply with the institutional and local requirements of each site, and it is each site’s responsibility to ensure that these are communicated to the sIRB prior to the review. Information regarding local institutional policies, requirements from other research committees, or compliance with local laws or cultures will be incorporated into the submission before it reaches the sIRB.
While the use of an sIRB is encouraged for NIH sponsored studies and is permitted under FDA regulations, the University of Iowa IRB may agree to act as a sIRB with non-federally or industry funded research on a case by case basis.
As the University of Iowa has standing agreements with some institutions dictating the requirements for shared IRB review between the institutions, investigators who wish to pursue using the University of Iowa IRB as the sIRB of multiple sites should contact the Human Subjects Office (HSO) External Review Coordinator prior to initiating the HawkIRB, in order to assess what agreements are needed. For more information on the External IRB Reliance process, review the HSO website under “Central & External IRBs.”
When an investigator at the University of Iowa participates in a multi-site trial with another institution serving as the sIRB, that creates what is known as a “relying” relationship. The University of Iowa relies on that other institution’s IRB to review the research. Requests to participate as a relying IRB should be submitted in HawkIRB, and are initially evaluated by the External IRB Coordinator. If there is an already established reliance relationship, question I.1 will reflect this IRB as an option and should be selected as part of the submission process.
Investigators should select “Other” for the IRB and in HawkIRB question I.1 if there has not been a reliance relationship developed, then enter the name of the sIRB in the free text field. Complete the abbreviated HawkIRB application, providing all pertinent and requested attachments. The University of Iowa IRB reserves the right to retain oversight of any multi-site study rather than relying on another IRB. Some reasons for this include:
- UI research team members having a financial interest involvement,
- Enrolling vulnerable subject populations, or
- An unusual research protocol
- Use of recombinant DNA (rDNA) - Protocols requiring Institutional Biosafety Committee review must remain with University of Iowa IRB oversight.
If an investigator would like to participate in a multi-site trial with the UI IRB serving as the sIRB, please contact the HSO to discuss how to prepare for serving as the lead IRB of record. Once that occurs, should submit a full IRB application in the HawkIRB system, requesting review by IRB-01 or -02, depending on the nature of the study.
The HSO and the University of Iowa IRBs are experienced in navigating the world of sIRBs. When considering the sIRB model, please contact the HSO early in the process. Email uirb-external@uiowa.edu, and we will help you begin the process.
What’s All the Fuss? External IRBs and the Single IRB of Record Models

By Martha Hedberg
In our December 2015 and June 2016 issues of IRB Connection, we introduced the innovative approach to reviewing and approving human subjects research – the Single IRB of record, or sIRB. This issue of Connection is dedicated exploring the new and evolving model of single IRB oversight.
For University of Iowa researchers, your local IRB is the board here at our institution. There are three boards that review different types of research depending on the department or location in which the research is conducted:
- IRB-01 (reviews Biomedical research)
- IRB-02 (reviews Behavioral/Social Science research)
- IRB-03 (reviews research conducted at the Iowa City VA Health Care System (VAHCS) or using VA resources)
An external IRB would be any other IRB that is not directly supported by the University of Iowa. An external IRB could be at another academic/research institution, part of a hospital or medical center, or a commercial enterprise such as Western IRB (WIRB) or Schulman IRB. With the exception of the UI IRB-03, all of these options can be a single IRB of record for a multi-site study. Research conducted at VAHCS or using VA resources may only be reviewed and approved by IRB-03; the sIRB model cannot be used when the lead study site is under IRB-03 purview.
In a sIRB model, one IRB extends research review and oversight to more than one research site. That IRB is called the “IRB of record” or “lead (or reviewing) IRB.” The IRBs at the other sites rely on that lead IRB and are called “relying IRBs.”
Requirements
Using an external IRB or serving as a lead IRB may be required by a sponsor for a multi-site study. It may also be used when a UI researcher collaborates with researchers at other institutions to conduct research at more than one site. Beginning May 27, 2017, National Institutes of Health will require the “Use of a sIRB” on all multisite, federally funded research projects using the same protocol.
The first thing that needs to be in place is an agreement between the relying and the lead IRBs. This can be in the form of a Master Agreement or contract that can be used for multiple studies, as in the case of the Great Plains Collaborative Consortium (GPC), or with a commercial IRB, such as WIRB. The agreement to rely on another IRB for a single study is called an IRB Reliance (or Authorization) Agreement. These documents establish the IRB-to-IRB relationship and outline the responsibilities of all parties – the study teams, the PIs at all sites, the lead IRB, the relying IRB, the institution, and so forth. The UI HRPP (Human Research Protection Program) does have an established template for use if one is not available.
When the initial agreement is formalized, the lead (or reviewing) IRB can begin receiving study materials. Once the study is approved by the lead IRB, the research can begin at all of the institutions that were part of the original approval. The sIRB model has been recommended in order to reduce the otherwise duplicative nature of multiple IRBs overseeing research at each site for a multi-site study. This model helps to streamline IRB approval and oversight of research, in particular with subsequent amendments and continuing reviews. If additional sites decide to participate, then typically a modification or amendment to the originally approved application will need to be submitted to the IRB providing research oversight.
When initiating a new sIRB reliance model, plan on some extra time to obtain IRB approval. Because the initial reliance agreement has to be negotiated and executed by the institutional officials at all participating sites, the agreement takes additional time and effort to finalize.
Using the sIRB Model at UI
The University of Iowa IRB and Human Subjects Office are fully equipped to help you either serve as the lead IRB of record if you are interested in establishing a multi-site study using the sIRB model or rely on another IRB to serve in this role if the UI Principal Investigator is not the lead PI.
The University of Iowa Human Research Protection Program (HRPP) will generally consider reliance on an external IRB if the two following conditions are met: 1) The IRB is registered with the Department of Health and Human Services(HHS). In accordance with the HHS regulations at 45 CFR part 46, subpart E, all IRBs are required to register with HHS if they will review human subjects research conducted or supported by HHS (use of federal funding) and could be designated under an assurance of compliance approved for federalwide use (i.e., an FWA) by OHRP (Office of Human Research Protections). 2) The UI HRPP also prefers to rely on other IRBs that have achieved accreditation issued by the Association for the Accreditation of Human Research Protection Programs (AAHRPP).
Any UI faculty or staff can request to use the sIRB model. Any sponsor can be involved in supporting this model. Later in this issue of IRB Connection you will read about federal guidelines from the National Institutes of Health regarding the use of the sIRB.
The Human Subjects Office (HSO) has been coordinating sIRB models for nearly a decade and a little over a year ago a staff member specializing in this area was added to address the increase in reliance requests. Investigators interested in beginning a new sIRB model should contact the HSO as soon as possible. Some federal funding agencies are now incorporating the sIRB model as a requirement for funding. Industry sponsors are beginning to implement this model as well.
Considerations
When submitting a proposal that involves relying on another IRB, UI investigators need to consider what are called “local context issues.” Local context issues are local laws, policies, considerations or other information known at the research site that must be made available to the IRB of record when a reliance model is used.
There are two scenarios which should be considered in addressing local context issues:
- When the University of Iowa research team is relying on an IRB of record outside of the UI. In order to ensure compliance with all local context issues, especially when the UI research team is relying on an external IRB of record, a submission is required in HawkIRB. After the local context issues have been affirmed, the UI study team will receive an “approval to submit” notice from the HSO. This means that the UI study team may now be added as a site with the IRB of record. If it is a commercial IRB, then the IRB application may be submitted to that IRB.
OR
- When the University of Iowa is the lead IRB (or IRB of record), the UI PI needs to be aware of the local context considerations of all of the relying research sites. This information should be submitted as part of the IRB application every time there is an addition of a new research site. The UI PI will remain responsible for ensuring the research project includes and addresses all local context issues. Researchers from participating (or relying sites) should be named as Non-UI Study Team Members in section II of the HawkIRB application. Ensure the HawkIRB application clearly describes activity as it occurs, at each site, for each question. If an activity will only occur at some sites but not others, this needs to be specified so the IRB can understand what activity is happening at each location. The UI IRB strongly recommends submitting each applicable research site individually via modification so each modification can individually consider the research site’s local context issues.
Information which may be considered as part of a “local context” review:
- State and local laws.
- Institutional Human Research Protection Program requirements. At the UI these could include, but are not limited to:
- If the study involves a drug at the UIHC, then review and approval by the Pharmacy and Therapeutics Committee would be required.
- Radiation Protection Committee
- Research Billing Compliance
- HIPAA
- Who will serve as the Privacy Board?
- Will the research site require use of its own HIPAA language or will the IRB of record require standard language?
- Compensation for Injury
- Are there specific contractual obligations required at the research site?
- Is there specified language the institution will require in the informed consent document?
- Other institutional or local policies that would affect the conduct of the research study? At the UI these could include, but are not limited to:
- The UIHC policy requirement for a record of consent.
- The use of a gift card for subject compensation requires the retention of the subject’s name and address or other applicable contact information.
- Mandatory reporting requirements
While it may seem a bit daunting and confusing initially, the staff of the HSO are well prepared to help you navigate this new approach to research review. Investigators may contact the Human Subjects Office at 335-6564 and ask to speak with someone about using the sIRB method. Extensive information is also posted on our website under the Central & External IRBs tab.
What Does It Mean to Rely on a Commercial IRB?

By Kathy Beck, CIP
University of Iowa researchers have the option to use a commercial (a for-profit business entity) IRB for review and oversight of new industry initiated and industry sponsored protocols that meet certain criteria. In general, this option is used if:
- The study involves a protocol designed and written by a sponsor that is a for-profit entity
- The sponsor holds the Investigational New Drug (IND) application(s) for the project,
- The project does not involve xenotransplantation, embryonic stem cells, or require review by the Institutional Biosafety Committee
- The study does not involve Iowa City VA Health Care System (VAHCS) resources or the recruitment of VAHCS patients.
The Initial IRB Application
If the University of Iowa has entered into a Master Service Agreement with the commercial IRB, the IRB will generally be included in a list of possible IRBs in Section I.1 of the HawkIRB New Project form. If the commercial IRB being requested is not listed as an option in HawkIRB, that means an IRB Reliance (or Authorization) Agreement needs to be negotiated. If that’s the case, the investigator chooses the “Other” IRB option and enters the name of the proposed external IRB in response to Section I.1.a. The University of Iowa reserves the right to withhold any research protocol, regardless of sponsorship or funding, from being sent to an external IRB for review. Research studies, including, but not limited to, the following require review by a University of Iowa IRB:
- Xenotransplantation
- Embryonic stem cells
- Review by the Institutional Biosafety Committee
- Research funds from a not-for-profit funding source
- Planned Emergency Research under FDA 21 CFR 50.24
The Pre-Project Checklist addresses the first four of the criteria above for the commercial IRBs listed in HawkIRB. If “Other” IRB is selected, these questions are integrated into the application.
Other 'Commercial' IRBs
When a UI investigator is approached by a sponsor to use a commercial IRB that is not currently listed in HawkIRB, they should (1) contact the Human Subjects Office and (2) submit the New Project application in HawkIRB so the reliance review process can begin. As per the UI Human Research Protection Program (HRPP) policy outlined in the External/Central IRB Reliance Process SOP, the UI HRPP Review Committee conducts a review of submitted materials to decide if the study may use an external IRB. If approval is granted, the negotiations to establish a reliance agreement begins. These negotiations can occur concurrently with the other UI committee reviews that must take place before the study can be submitted to the external IRB.
Attachments
When selecting the “Other” IRB option, the Assurance Document generated from the Attachments page of the HawkIRB application will be automatically populated with “Other IRB” as the IRB of Record. The investigator will need to revise this document to indicate the name of the specific IRB. The investigator must attach the template Informed Consent Document, study protocol, and signed Assurance Document before submitting the HawkIRB New Project form.
Informed Consent Document
Similar to what has been outlined in the above article titled “What’s All the Fuss? External IRBs and the Single IRB of Record Models,” Commercial IRBs are also required to consider local context issues. The informed consent language is one of those areas. There are several sections of the Informed Consent Document that use standard language for all UI research, regardless of the IRB of Record. Agreements about privacy protections for the use and creation of medical records (HIPAA), plans for compensating subjects for research-related injury, and who will act as the privacy board are all negotiated between the Commercial IRB and the University of Iowa. The UI investigator may need to revise the HawkIRB application and consent document(s) once the agreement is finalized.
The HIPAA and Compensation for Injury sections of the consent document must use UI template language. The required template language can be downloaded in the attachment section of the application. Additionally, the UI IRB expects that the consent document will provide subjects with the “name or other specific identification of the person(s) or class of persons” who will have access to their health information and to clarify what info is being provided and why, as per 45 CFR 164.508 (c)(iii). Our preference is always to name specific persons or entities. However, terms that represent a contractual or fiduciary obligation to a sponsor may be accepted. Entities listed in other areas of the consent, such as a confidentiality section, should be consistent with the entities listed within the HIPAA section.
There may be additional required language to meet Human Research Protection Program (HRPP) committee requests and to comply with institutional, University of Iowa or University of Iowa Hospitals and Clinics policies, Iowa State Law requirements, and other federal requirements. Examples, of required language include, but are not limited to: CMS/Medicare language, Clinical Trials.Gov reporting, Collection and Use of Social Security Numbers (SSN), Pregnancy testing for females under the age of 18 and Legally Authorized Representative language.
Essential submission steps for a study relying on a commercial IRB:
- Initiate a HawkIRB New Project application, which includes a pre project form (or eligibility review) checklist.
a.The pre project form determines if a research study meets eligibility requirements to be sent to a commercial IRB. (as noted earlier in this article) - If eligible: a banner will open, indicating this study may be submitted to an external IRB. If the study does not meet the initial screening criteria, a banner will open instructing you to submit to the local (UI) IRB. Select the applicable IRB in section I.1 in the HawkIRB application.
- Complete and submit the HawkIRB application
- Please see the Human Subjects Office website for more details on External IRBs and submission instructions.
- The IRB of Record (Commercial IRB) will provide the approval documents to the investigator after all of the HRPP committee approvals are in place.
Tracking of the research occuring at the University of Iowa
The University of Iowa research team will be responsible for adhering to all IRB requirements of the IRB of record, all University of Iowa institutional policies and procedures, and all Human Research Protection Program Committee(s) policies and procedures. An application is submitted in HawkIRB application to gather information for these required reviews. The HawkIRB submission serves as institutional documentation of the ongoing research. Under the Federalwide Assurance (FWA) filed with the Office of Human Research Protections, the University of Iowa remains responsible for the conduct of the research regardless of who serves as the IRB of record. The UI researcher should submit a HawkIRB application concurrently with the application to the external, IRB of Record.
Post Approval Activities
After the Commercial IRB approves the study, the UI researcher must submit modifications, continuing reviews, reportable events, and closure requests to the external IRB of Record and concurrently in HawkIRB. Modification forms describe any changes in the study design and procedures. At least once a year, the UI researcher must submit a Continuing Review form if the study is still enrolling or collecting data from subjects or working with identifiable data. Reportable Event Forms (REFs) are to be submitted in HawkIRB when submitted to the external, IRB of Record. A Project Closure Form needs to be submitted in HawkIRB, in addition to the closure documentation required by the IRB of Record. The UI IRB reviews these submissions concurrently with the Commercial IRB of Record. Commercial IRBs follow the same general practices as WIRB. Additional information about protocol modifications and other post-approval submissions for all Commercial IRBs can be currently be found on the Human Subjects Office website.
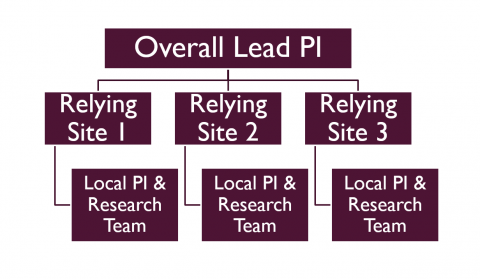
PI Responsibilities when utilizing a Single IRB
by Anna Mertes,, M.ed
Responsibilities for the Lead Site Investigator

If you are the overall lead study Principal Investigator (PI), it is important to know the expectations when engaging in a multi-site research project utilizing a single IRB. It is your responsibility to know the expectations of both the lead and relying sites which will be outlined in the Agreement and the Standard Operating Procedures manual (if applicable). A few examples of information you will need to know as the Lead PI include:
- The state or local laws and institutional policies that impact how a protocol will be implemented for each site.
- How you will train study personnel across sites
- How you will communicate across sites
- How you will monitor compliance
- How you will address and report unanticipated problems or noncompliance
- How you will identify and report to the IRB all reportable events
As part of the reliance process, the IRB of Record will ask the relying sites to inform them of any local context considerations the IRB of Record should know while reviewing the application. These local context considerations include local/state laws, institutional policies, training, and HRPP committee reviews (DSP, P&T, JOC, NRC, etc). In terms of how you will communicate across sites, developing a communication plan will be crucial. A communication plan will need to be included with your NIH grant submission, so we encourage to consider this early on in your process. An easy way to think about it is to consider each study site as an extension of your local research team. Your communication plan with all study sites should include:
- a regular site to site communication plan
- study specific training
- some examples could include training outlining conduct of the research, specific aspects of the protocol, how to share research related information, how to use data collection forms, etc.
- dissemination of new information which may impact the conduct of the study or the subjects’ willingness to continue participation in the research
- addressing adverse or reportable events,
- compliance concerns,
- who and how to contact you with questions, etc.
In terms of the reportable events and serious or continuing noncompliance concerns, many sites have their own reporting obligations in addition to what needs to be reported to the IRB of Record. When these situations arise, be sure both the local IRB and the IRB of Record are informed of the situation(s). Here at the University of Iowa, you can inform the IRB of any noncompliance concerns by completing the REF form in HawkIRB.
It is also important to know you are responsible for collecting information and reporting it to the IRB of Record for each relying site. This reporting obligation relates to modifications, continuing reviews, and reportable events that occur for each site. You will also be responsible for maintaining and submitting the paperwork for each of the relying sites. For example, if you have three relying sites, it is likely you will maintain three versions of the informed consent, recruitment materials, delegation of authority logs, etc. As you can imagine, the role and responsibilities of the lead PI and the lead IRB are quite vast.
Responsibilities for the Relying Site Investigator
If you are the PI at a relying site, you have responsibilities to both the local and reviewing IRB. The local UI HRPP (administered by the HSO) is responsible for conducting the HRPP review regardless of who serves as the IRB of record. This means you will submit application materials to the UI IRB for the initial review and for any modifications, continuing reviews or reportable events for the duration of the study. However, the IRB of Record is responsible for providing the regulatory oversight for the research protocol. So they will provide the IRB review for the initial review, and any modifications, continuing reviews, or reportable events for the duration of the study. A few examples of information you will need to know as a relying site PI include:
- What does the IRB of Record require?
- How will I find out the IRB of record policies?
- How do I communicate local context issues?
- How do I (or will you) submit to the IRB of Record?
- How will I receive my IRB approved documents?
- Who do I notify if there are problems or reportable events?
As a relying site, the lead research team should be able to provide you with the information you need regarding the rules and expectations for the IRB of Record. As a relying site, you should have answers to all of the questions noted above in the “Responsibilities for the Lead Site Investigator” section.
Additionally, some institutions post their policies publicly on their institutional website. If the research project is utilizing a Master Agreement, you may also have a Standard Operating Procedures (SOP) document you can reference when you have questions. If an SOP is not available, a great resource is the PI of the site where the IRB of Record is housed. If the PI cannot assist you, please reach out to me and I will do my best to help you find the answers you are looking for.
First, and foremost, when you are considering participating in a multi-site research project, please contact the Human Subjects Office. We understand this process can seem daunting, but hopefully it will reassure you to know we have participated in multi-site research projects utilizing a single IRB in both the lead and relying site roles. As the External IRB Coordinator, I am here to help you navigate this process. If you have any questions or are considering a multi-site project, please contact me by email at uirb-external@uiowa.edu or by phone at 319-335-9915.
Single IRB Q&A Info Spot:

By Kathy Beck
Beginning in January, 2017, we will have a standing section in the IRB Connection Newsletter dedicated announcements and topics related to the use of external IRBs. This will include information on either relying on an outside IRB of record, using a commercial IRB, or serving as the lead PI when the University of Iowa IRB may be the IRB of record. Please send your questions or topic suggestions you would like to see highlighted to uirb-external@uiowa.edu or UIWIRB@uiowa.edu or call 319-335-7297. If you need an immediate response to a question and cannot locate an answer on the HSO website in our “Central & External IRB" section, please contact me.
Herky Hints: UI IRB Reliance Options and HawkIRB

By Sarah Heady
In Brief
As noted in this IRB Connection topical series, UI researchers can now take advantage of increasing IRB review options in HawkIRB. Because IRBs other than the University of Iowa IRBs (IRB-01 and IRB-02) may be the lead IRB for all sites involved in a research project, HawkIRB provides different methods to describe variations in IRB review. If the UI is the IRB of Record (approving the research for all sites) the HawkIRB application must include details and variation for each and every research location. If the UI agrees to rely on another IRB to act as the IRB of Record, much of the HawkIRB application will close. (Think of this as ‘HawkIRB Lite.’)
Note: VA research cannot use the external IRB model or reliance model so the information presented in this article does not apply to HawkIRB applications for IRB-03.
When the UI is the IRB of Record
If other sites will rely on the University of Iowa as the IRB, the HawkIRB application should describe the research activity at each site. If research activities will vary from site to site, these variations should be described clearly throughout the application. This includes listing all research team members at all sites, stating all locations where study procedures will occur, and describing all possible methods for recruitment, obtaining consent and conducting study procedures. Also be sure to address any local context issues the IRB may need to consider in their review.
Research Team Members
All research team members at each site must be listed in Section II of the application if they are engaged in human subjects research. Non-UI team members are added just below UI team members—check the box to add non-UI research team members. You will need to provide complete information for all non-UI team members, including an e-mail address, name of the institution, and city/state location. If an institution has a Federalwide Assurance (FWA), you will need to provide that number as well. A brief description of the individuals research related activities should be included in the section noted “role” so the IRB can determine whether or not each non UI research team member meets the regulatory definition of “engaged in research.”
Conflict of Interest and Research Team Members
Other issues that must be considered when adding research team members: are there personal, professional, or institutional conflicts of interest that require disclosure? If an Individual Investigator Agreement is signed, the UI will request disclosures from those individual/s. If a Reliance Agreement is used, the relying site (the site relying on the UI IRB) will need to submit information about any potential conflicts of interest.
Key Personnel
Key personnel are team members who are able to make independent decisions about the research design, protocol, and subsequent outcome analysis. HawkIRB automatically assigns the PI and any faculty members as key personnel; non-UI research team members may to discuss their roles with the Conflict of Interest in Research Office at 4-4256 or coi-research@uiowa.edu
Subjects
Section VI of the HawkIRB application is the spot to list inclusion and exclusion criteria, demographic estimates of potential available subjects, and many other details related to subject populations. This is another area of the application where differences between sites must be carefully described. Perhaps the local UI site will enroll minors, and the relying sites will only enroll adult subjects—Section VI is the area where this should be clarified.
Study Location and UI Role in the Project
In Section VII.A.1, make sure to list all locations where research and research activities (including data analysis) will take place, both on and off the UI campus. As with all other questions, if you run out of space in the text box, attach a document with the locations listed, and reference the name of the document in the response. When the UI IRB is the IRB of Record, make sure to select ‘Coordinating Center’ along with other applicable options (e.g. clinical/participating site, central laboratory, etc.). VII.A 4 will open a text box requesting more information. Pay special attention to capturing the details about the UI leadership role—how will all of the variations and updates in procedures and protocols be communicated, and how will adverse events be reported and handled? When the UI IRB takes on the role of the IRB of Record, all aspects of the research project at each site location must be completely described in the application responses.
Recruitment and Consent
In section VII.D, be sure to capture how recruitment and consent may differ from site to site—some locations may only analyze specimens for example, and may not be involved in consent and recruitment. All of these differences should be described. If the UI IRB is serving as the privacy board for each site, questions VII.D.2 – VII.D.6 must address HIPAA compliance for each location.
Study Procedures
As with recruitment and consent, responses in Section VII.E should describe all variations in research procedures and study follow-up for each location.
Attachments
In addition to application responses, all documents regarding the study must be attached to the application—protocols, consent documents, recruitment materials. The IRB of Record must review and approve the documents used at all study sites. This may involve some back and forth negotiations with project sponsors and/or funding agencies while finalizing informed consent documents, in particular. Some sections of the Informed Consent Documents may be edited; typically, the HIPAA information and the details about compensation for injury should reflect local context language. (Don’t forget to include all variations of recruitment posters or brochures that will be used at each of the sites.)
When Another IRB Oversees the Study
If the UI IRB agrees to rely on an external IRB as the IRB of Record, the HawkIRB application is condensed to the essentials that cover what the UI Human Research Protection Program (HRPP) needs to know about the UI site. As noted in earlier articles, the HawkIRB submission fulfills institutional documentation requirements of the ongoing research. Under the Federalwide Assurance (FWA) filed with the Office of Human Research Protections, the University of Iowa remains responsible for the conduct of the research regardless of who serves as the IRB of record.
The application should still capture relevant details. And please note that all Other Committees that are part of the UI Human Research Protections Program will still use the HawkIRB application to conduct their review. For example, studies of an investigational drug must be reviewed and approved by the Pharmacy and Therapeutics (P&T) Committee and Investigational Drug Services (IDS). The PI will need to generate the Drug Charge Worksheet and the Drug Study Form, complete the forms, and attach them to the HawkIRB application. The PI may need to respond to comments from IDS and make changes to drug handling, dispensing, or documentation. The study will not be released until all committees sign off on approval of the project.
After Approval: Modifications, Continuing Reviews, and Reportable Events
Responsibilities don’t end when the project is approved—even when UI IRBs rely on another site as the lead IRB, Principal Investigators are still responsible for keeping the HawkIRB application up to date with any changes by submitting modifications in HawkIRB. In general, UI PIs should submit to HawkIRB first, and then to the lead IRB. For example, research team additions at the University must meet UI requirements—this needs to be documented before the lead IRB signs off on the approval modification.
Continuing Reviews should always reflect information about the progress of the project at the UI site—you do not need to report total cumulative enrollment for a multi-site project, but the enrollment numbers for all UI subjects should be reported. Attach any relevant documentation submitted to the lead IRB—for example, funding or progress reports.
Adverse events involving UI subjects, or subjects enrolled by a UI investigator, should be reported in HawkIRB. Adverse events always need to be reported to the lead IRB. How they are reported will depend on the type of event and the IRB policies of the lead site. Be sure to consider how reporting requirements may vary by site, and plan to communicate with the HSO and the lead IRB site if you have questions.
Establishing Trust in Partnerships is Key to Centralized IRB Review

By Brent Collinsworth
According to Cherie Bilbie, one of the most important components of the centralized IRB review process will not appear on any contract or database. Ms. Bilbie, the director of the human research protection program at Hartford HealthCare in Hartford, CT, states that the key to effective central/external IRB functioning is trust and close relationships between researchers, staff, and other IRBs. A core way to build trust in this process is through organization and clear expectations. Ms. Bilbie states that template agreements, documents that clearly delineate responsibilities and expectations to multiple IRBs in a multi-site study, can be a key way for everyone in the review process to know their part. The Hartford HealthCare IRB also has an electronic IRB system that is able to serve as a repository for research-related documents and information. This allows yet another avenue for keeping everyone on the same page and ensuring compliance.
Read the full article in the November issue of IRB Advisor. The November issue also includes articles about:
- The NIH/FDA clinical trials reporting rules and how they will expand clinical trials transparency
- Highlights from the NIH final rule on clinical trials submissions
- A recently published study that shows the close ties between researchers studying the health effects of sugar and the sugar lobbies in the 1960s
- An interview with author and professor Dr. Marion Nestle about the sugar lobby’s ties to research
- The BEAM Program, a mentoring program for careers in human research ethics focused on youth and young adults
- Discussion about whether IRBs should require researchers to disclose assessments of research reproducibility in their protocols
What is IRB Advisor?
IRB Advisor is a monthly newsletter that contains articles about regulatory issues, informed consent, current events in human subjects protections as well as articles about IRB administrative and management issues. The UI IRB subscribes to this publication as a resource for UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff. Each month the IRB Connection Newsletter features an article from the current issue of IRB Advisor.
Current and Past Issues
There is a link to current and past issues of IRB Advisor on the Education and Training page of the Human Subjects Office web site. This link provides automatic access to the newsletter from all computers with a University of Iowa IP address.
The University of Iowa username and password cannot be posted on the Human Subjects Office web site. UI faculty, staff and students and VA researchers may contact the Human Subjects Office to request the username and password to access IRB Advisor from a personal computer. Contact us by e-mail (irb-outreach@uiowa.edu) or call us at 319-335-6564.
Continuing Education Credits
Individual newsletter subscribers can receive 1.5 AMA PRA Category 1 Credits™ or 1.5 nursing contact hours for reading an issue and completing an online test. However, since this is an institution-level subscription, UI researchers must purchase access to an individual issue for $40, or purchase a full subscription, to receive CME/CE credits. Visit the AHC Media web site for information about subscription options.

