Index
Announcing Human Subjects Office (HSO) Education and Compliance Openings
Reverting to Virtual Presentations Only Beginning November 1
WIRB / WCG Online Submission Platform Training November 15th
Hot Topics & IRB/HRPP Updates
Research Billing Compliance: Ensuring a smooth road to success for your study
An Overview of the IRB Review Process
Locating Support and Training Materials for CT.gov
In the News
Announcing Human Subjects Office (HSO) Education and Compliance Openings
The HSO seeks applicants for several Compliance and Education Specialist positions. The Compliance & Education Specialist (PRC1) or Senior Compliance & Education Specialist (PRC2) are both Professional Scientific positions. To express interest at either level, please apply to requisition 21004074 or requisition 21004905 at https://jobs.uiowa.edu.
The Compliance & Education Specialist (PRC1) will provide developmental and administrative assistance and expertise for the human subjects protection programs and services administered by the HSO. This position will schedule, prepare, promote and present educational activities for the purpose of compliance with federal regulations, IRB requirements, and University of Iowa policies and procedures for the conduct of research involving human subjects.
The Senior Compliance & Education Specialist(s) (PRC2) will act as education and compliance expert regarding federal regulations, Institutional Review Board (IRB) requirements, and University of Iowa policies and procedures for the protection of human subjects involved in research. Responsibilities will include to develop and coordinate procedures and programs to enhance investigator understanding of the HawkIRB application process and compliance with the Institutional Review Board (IRB) policies and procedures. Occasional in-state and out-of-state travel is required.
These positions are open to a full time equivalent of 50-100%. Part-time or full-time appointments are available within the stated range. Hybrid remote or telework schedules will be considered.
Applicable background checks will be conducted on all positions. The University of Iowa is an equal opportunity/affirmative action employer. All qualified applicants are encouraged to apply and will receive consideration for employment free from discrimination on the basis of race, creed, color, national origin, age, sex, pregnancy, sexual orientation, gender identity, genetic information, religion, associational preference, status as a qualified individual with a disability, or status as a protected veteran.
For more information, please see: https://research.uiowa.edu/office-vice-president-research/open-positions
Reverting to Virtual Presentations Only Beginning November 1
The Human Subjects Office (HSO) began providing IRB presentations and HawkIRB trainings in a hybrid format (in-person and via Zoom) at the start of the Fall semester in August. Due to the very low in-person attendance rate and consideration of the continued health of the UI research community going into the flu season, the HSO has elected to return to an all virtual format for presentations and trainings beginning on Monday, November 1, 2021.
If you registered to attend an IRB presentation or a HawkIRB training after November 1st, there is no action required. The Zoom link was in the confirmation email and calendar invite that you received. However, if you indicated in the registration survey that you would attend in-person, you should attend virtually instead.
Visit the Education and Training page of the HSO website for a list of the remaining IRB presentations and HawkIRB trainings for this semester. If you have any questions or would like to confirm your registration for a presentation or training, please contact the Education and Outreach team.

WIRB / WCG Online Submission Platform Training November 15th

[EDIT: This presentation was cancelled due to weather and rescheduled for December 8th]
On November 15th, 2021 at 3:00 pm CST, the Human Subjects Office (HSO) will facilitate a virtual training to help research teams become familiar with Western Copernicus Group's (WCG) new online submission system. In this one-hour training, a representative from WCG will provide an overview of the new WCG Connexus online portal, as well as insight and guidance for what to expect from this system.
Pre-registration is required. Once you are registered, the HSO will send you an email with the Zoom link and calendar invite. If you have questions, please feel free to contact Mayzie M. Tucker, Commercial IRB Application Analyst.
Hot Topics & IRB/HRPP Updates
By Kelly O'Berry, BS, CIP
This article provides an overview of the topics covered in the October 2021 presentation about hot topics and updates from the Institutional Review Board (IRB) and the Human Research Protection Program (HRPP). A recording of this presentation is available in the Additional Topics section of the IRB ICON Course for Researchers.
Data Governance Committee Policy/Procedures for Sharing Data Outside UIHC

Boyd Knosp, Associate Director of Informatics in the Institute for Clinical and Translational Science (ICTS) shared information about the steps in the process of sharing data from the UI Health Care source to other researchers. This process is overseen by the UIHC Data Governance Committee. Data governance refers broadly to how organizations make decisions about data. For sites like the University of Iowa that have Clinical and Translational Science Award (CTSA), there is a growing interest and focus on data governance.
The UIHC Data Governance Committee reviews the use of operational, clinical and research data from UI Health Care. Research data could be shared outside the UI Health Care covered entity to a UI collaborator, outside the UI to a non-commercial recipient or outside the UI for commercial use. Boyd discussed the factors the committee considers related to sharing research data and the process for obtaining approval.
UIHC Researchers who want to share data should contact the Data Governance Committee at icts-bmi-consulting@healthcare.uiowa.edu. To avoid unnecessary delays, the HSO/IRB strongly advises working with the ICTS BMI Consulting team to obtain Data Governance Committee approval prior to the submission of an IRB application. If a HawkIRB application indicates plans to share data outside UIHC, the HSO/IRB staff reviewer may ask the researcher to obtain Data Governance Committee approval.
UI and IRB Policies/Procedures for PIs Leaving and Taking Data
Principal Investigators should plan ahead to take care of their HawkIRB applications and obtain permission to take data with them. At least three months before they leave, the PI should complete the PI Transfer/Departure Checklist (for federally-funded research) or a PI Transfer/Departure Checklist (for federal and non-federal sponsored projects (funds 500/510)) or an Internal/Unfunded PI Transfer/Departure Checklist. Additional information regarding PI departures can be found on the Division of Sponsored Programs (DSP) website. A Human Research Protection Program (HRPP) Departure Checklist is also available as a checklist to ensure human subjects research processes are all addressed prior to a PI’s departure..
The PI must decide what will happen with their study after they leave. They need to submit a Modification form to change the PI if another researcher will continue to oversee the research at the UI. If the HawkIRB application does not already indicate in Section X.5-6 that the PI will take data with them, that will need to be modified as well. For studies involving prospective data collection, the IRB can only approve this if it is consistent with what subjects agreed to in the Informed Consent Document.
The PI must also make plans to comply with the UI record storage requirements. The UI IRB policy for record storage is found in the UI IRB Standard Operating Procedures and Researcher Guide, Section II, Part 23 (Record Retention Requirements). This policy only applies to:
- Copies of the human subjects research application forms
- Notices of IRB approval
- Signed Informed Consent Forms
In order to take data with them, the PI may need a Data Use Agreement and/or Materials Transfer Agreement. Both agreements require institutional and departmental approval. The PI should work with the Division of Sponsored Programs (DSP) to establish these agreements. DSP will verify that the PI has IRB approval to take data with them.
IRB Educational Tools
The Human Subjects Office creates Educational Tools to provide more in-depth guidance on various topics. These tools posted on the Education and Training page of the Human Subjects Office website. Many of the nine current tools were created in collaboration with, or at the suggestion of, a member of the UI research community. We invite suggestions for additional Educational Tool topics. Send them to IRB@uiowa.edu.
Exempt Application Form Updates & Upcoming Presentations
We completed the programming and started beta testing the new Exempt Application form since we discussed this initiative at the July 2021 Hot Topics and Updates presentation. The goal of this project is to reduce regulatory burden for researchers and for the IRB. The revised HawkIRB form will streamline the application form and submission process for research that qualifies for Exempt Status. It will also have more yes/no questions and checkboxes and fewer open text fields.
There will be three presentations this fall on the Exempt Application Form:
- Wednesday, November 3 from 1:00-2:00 pm – An overview of the HawkIRB revisions (via Zoom)
- Wednesday, November 10 from 9:30-10:30 am – An overview of the HawkIRB revisions (via Zoom)
- Monday, December 6 from 1:30-3:00 pm – A HawkIRB training specifically about the new questions and sections in the Exempt Application form (via Zoom)
We are still seeking volunteers from the UI research community, especially social/behavioral researchers, to help with live testing in December and January. Our goal is to roll out the Exempt Application Form before the beginning of spring semester on January 18, 2022. To volunteer, please send an email to IRB@uiowa.edu.
Future Hot Topics and HRPP Updates
Hot Topics and IRB/HRPP Updates lectures occur three times a year (July, October and February/March) to share information with the UI research community about common questions, new initiatives and HawkIRB enhancements. We welcome your suggestions for topics to include in future Hot Topics and IRB/HRPP Updates presentations. Please send your ideas to IRB@uiowa.edu.
Research Billing Compliance: Ensuring a smooth road to success for your study
By Denise Krutzfeldt, BA, CHRC, CPC-A
Compliance Associate Director, Patient Financial Services
Researchers submitting a New Project application in HawkIRB must obtain a number of Other Committee reviews along with IRB review, depending on the type of research they’re proposing. One of those Other Committees is Research Billing Compliance (RBC), whose purpose is to help ensure charges for clinical trial services at UI Health Care are billed in compliance with applicable regulations and policies, and to provide a centralized research billing process. Non-compliant billing can lead to serious consequences, including large fines, damage to the institution’s reputation, and even criminal penalties.
Research team members would agree that they have a strong interest in getting their study through the RBC approval process as quickly as possible. In addition, they would also agree they don’t want their study subjects’ insurance to be billed for services that are also being paid for or provided by the study sponsor (double-billing), or billed for services that were performed for research purposes only and were not part of the subjects’ clinical care. So how can a research team help make sure these aims are met?
Before IRB approval, RBC thoroughly reviews and analyzes the IRB submission, including the study protocol, consent document, and HawkIRB application, along with the sponsor agreement. Whether the study is a Medicare Qualifying Clinical Trial, what the study sponsor is paying for or providing, and what the consent document says are all taken into account by RBC during the process of building the Research Billing Plan. Consistency and accuracy throughout the study documents and prompt replies to questions keep the RBC approval process moving, including a speedier turnaround of the final Research Billing Plan that is sent for the Principal Investigator to review and sign.
After IRB approval, timely association of enrolled subjects to the study in Epic is crucial in making the “back end” part of the process work. This step is the only way to stop and hold the subjects’ charges in Epic for RBC review; without it, the charges can be billed to insurance, including charges for services that are being provided for or paid by the study sponsor or shouldn’t be billed to insurance. RBC relies on the research team to associate all study subjects timely. In addition, the process of reviewing charges in Epic inevitably leads to questions for the research team, even when RBC uses the Research Billing Plan as a guide. Prompt responses to RBC’s questions ensure the subjects’ charges are not held in the revenue cycle.
In sum, the following tips will help your study run smoothly from a Research Billing perspective, both during the IRB approval process and when the study begins:
- During the HSO/IRB/Other Committee review process:
- Make sure the IRB submission is consistent in describing the study procedures and who is paying for what, including the HawkIRB application, study protocol, consent document, clinical trial agreement, study budget, etc.
- Answer questions from RBC staff promptly as they conduct the Coverage Analysis and develop the Research Billing Plan
- Review the final draft of the Research Billing Plan before the PI signs it, then return it promptly to RBC
- After IRB approval and the study begins:
- Associate subjects to the study in Epic in a timely manner; specifically, at the time the subject signs the consent document and before study procedures begin
- Answer questions from RBC staff promptly as they do the Epic charge review
- Notify RBC of unexpected subject visits or charges
- Notify RBC if you submit a Modification that would affect the Research Billing Plan (more or fewer study visits or study procedures; change in the PI, etc.)
For more detailed information see the Research Billing Compliance page on The Point (the intranet site for UI Health Care employees). Questions regarding policy and/or procedures can be directed to the Research Billing Compliance at: researchbilling@healthcare.uiowa.edu or
via phone (see phone directory The Point).
An Overview of the IRB Review Process
By Joanie Hoefer, BS CIP
You have an important research question to answer. You worked diligently on filling out the HawkIRB application and even attended HawkIRB trainings and Office Hours to make sure you were doing everything correctly. The much-anticipated moment comes when you hit the button to submit your form in HawkIRB. Now what? What is the Human Subjects Office (HSO) and the Institutional Review Board (IRB) doing with that form that you just submitted? This article will describe what generally happens during the IRB review process to give you an idea of how your forms are handled and reviewed once they have been submitted.
Step 1: Administrative review

The first step for all HawkIRB forms is an administrative review by an HSO staff reviewer (this is also called “pre-screening”). This is the initial check of the application to make sure the PI selected the correct IRB, attached all expected documents to the form and reasonably answered all questions.
If it is a New Project form or a Modification form to change the Principal Investigator (PI), the Assurance Document is thoroughly reviewed. The staff reviewer makes sure all required signatures are provided and that they are accompanied by a printed name. Electronic signatures are only accepted if the signature can be verified through an authentication feature.
The reviewer then looks at the responses to the form questions. They will note any potential issues with the form and then send it on to an Application Analyst.
Step 2: Application Analysis
Application analysis is a high level of review and involves a complete analysis of the responses and attachments provided in the form. During this level of review, the Application Analyst makes requests or suggestions for revisions to the HawkIRB application. Staff reviewers make sure that there are consistent, detailed answers to each of the application questions so that the IRB Chair or the full board has a complete and accurate description of the study for their review. Many common concerns of the IRB Chairs and/or the full board are also addressed during this step of the review process. Application Analysts make sure the HawkIRB application fully addresses the regulatory criteria for human subjects research approval before the application is sent to a Chair or the board for review. Those criteria include:
- Risks to subjects are minimized.
- Risks to subjects are reasonable in relation to anticipated benefits.
- Subject selection is equitable.
- Informed consent is sought from the subject or their legally authorized representative.
- Informed consent is documented or appropriately waived.
- The research has an adequate plan to monitor subject safety when it is appropriate.
- Adequate provisions to protect subject privacy and the confidentiality of the data are in place.
- Additional safeguards for vulnerable subject populations are included.
During the application analysis step, staff reviewers and others involved in the Human Research Protection Program (HRPP) are also reviewing information in the HawkIRB application to ensure consistency with state, local, other federal, and institutional requirements. These reviews generally occur concurrent with the staff and IRB review. Information on the other research review processes can be found on the HSO website or in the UI SOP and Researcher Guide (Section I; Parts 2, 3, 8, and 9A)
Step 3: Final IRB Chair or full board review
Whether your application is reviewed by a single IRB Chair or the full board during Step 3 is dependent upon the level of risk the study poses to subjects. If your study involves no more risk than what the participants are exposed to during their day-to-day life (i.e. minimal to no risk) and is eligible for either expedited review or exempt status, then Step 3 will be completed by a single IRB Chair or Chair Designee (a board member designated by the IRB Chair to conduct these reviews). However, if an IRB Chair is uncomfortable approving a study due to some aspect of the study, (e.g. controversial topic, potential risks to the researcher or liability to the University, etc.) they may opt to send the study to a convened board meeting for review and approval. If the study involves greater than minimal risk to the subjects, the final review of your application will be completed by a convened IRB board.
For additional information about the IRB review process, review the Frequently Asked Questions topics What to Expect During the IRB Review Process and the Status of a Submitted Application on the HSO website or Section I, Part 11.C in the University of Iowa IRB Standard Operating procedures (SOP) & Researcher Guide. In a future article, we will describe how long the review process typically takes and factors that can shorten or lengthen the review time.
Locating Support and Training Materials for CT.gov
By Fozia Ghafoor, MBBS
Researchers are required to register all applicable clinical trials (ACT) on the ClinicalTrials.gov (CT.gov) Protocol Registration and Results System (PRS). The CT.gov PRS is a web-based data entry system used to register a clinical study or submit results information for a registered study. There are many resources available to help researchers learn about CT.gov requirements such as investigator and user guides, the HSO website and tutorials, support and training materials on the CT.gov website. This article will give an overview of these resources that can help researchers understand the basic steps of the registration process and results data entry.
CT.gov establishes one PRS account per organization. Each PRS account is managed by one or more institutional administrators who can add an unlimited number of user logins. The University of Iowa (UI) has a designated PRS administrator who manages the institution’s account and creates logins for additional UI users. Researchers must have a PRS account to register study information on CT.gov. If researchers do not have an account, see How to Apply for a PRS Account. You can reach the PRS administrator at ct-gov@uiowa.edu. Once the account has been created, you will receive an email with instructions for logging in to the PRS.
Locating Support and Training Materials on ClinicalTrials.gov
Support materials
Support materials can be assessed by clicking on the “Submit Studies” tab at the top of the ClinicalTrials.gov website:
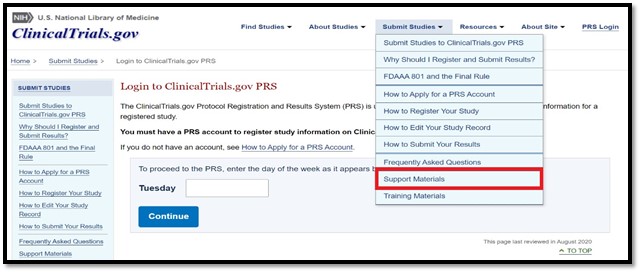
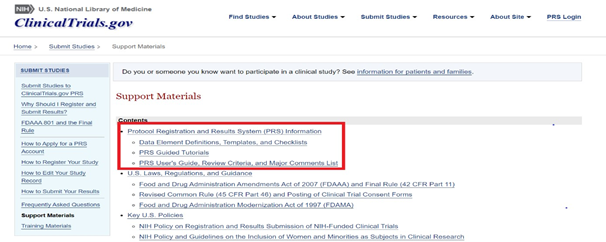
The “Protocol Registration and Results System (PRS) Information” link provides the definitions for protocol registration data elements submitted to CT.gov for interventional studies (clinical trials) and observational studies. These definitions are mostly adapted from the Final Rule. Data element entries are annotated with symbols to indicate generally what information is required to be submitted (and under which circumstances).
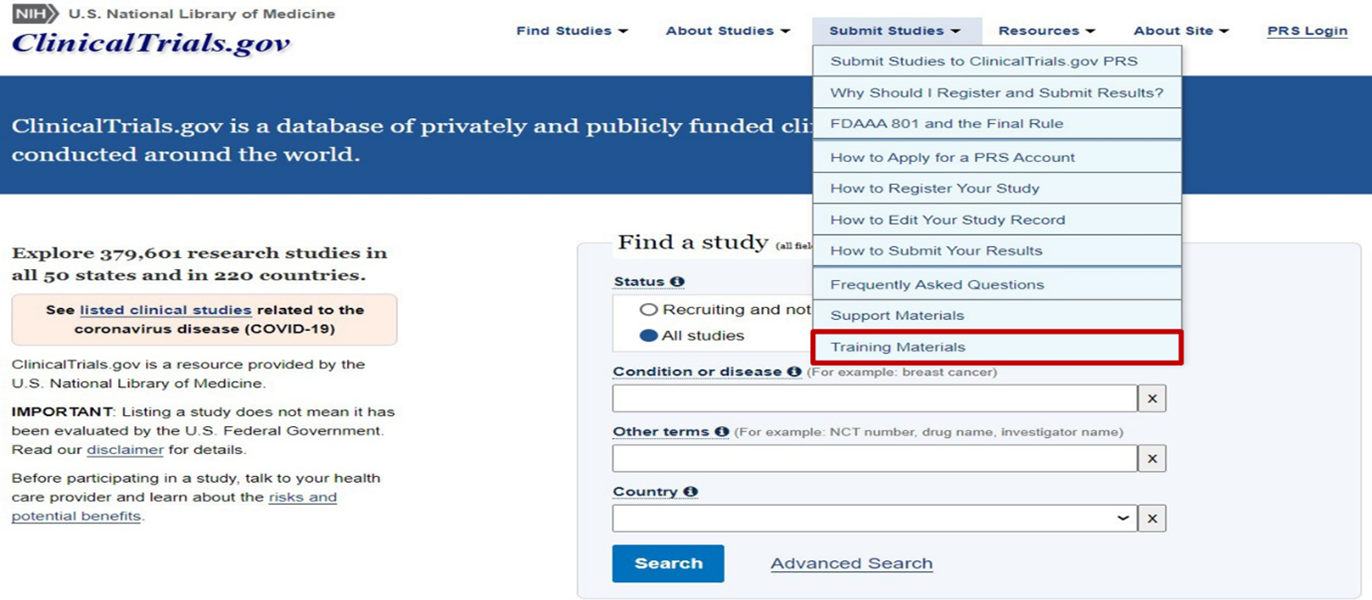
Training Materials
ClinicalTrials.gov staff has developed online presentations to help researchers during the study registration process and the submission of study results to CT.gov. These presentations can be found by first clicking on the “Submit Studies” tab and then selecting “Training Materials”:
In addition, the ”Example Studies for Results Data Entry” link has been created to illustrate key concepts for results data entry in the PRS.
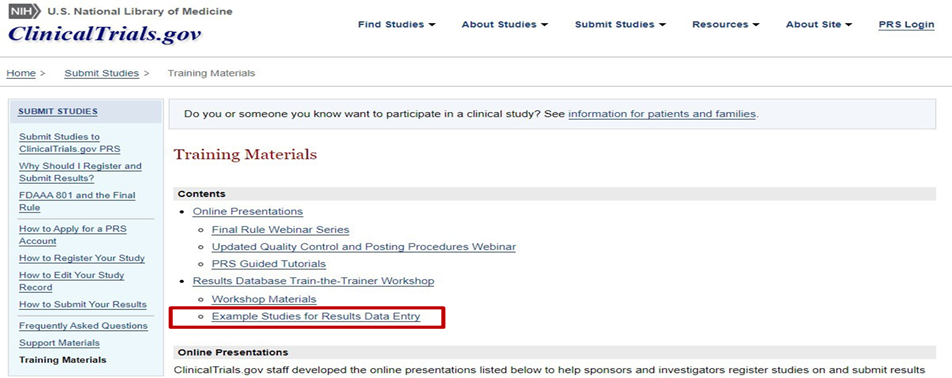
Examples of study records and papers for fictional studies were developed for the following various study designs:
- Parallel Study Design
- Cross-over Study Design
- Dose Escalation Study Design
- Factorial Study Design
- Multiple Period Study Design
- Units Other Than Participants
- Cluster Randomized Design
- Fractional Factorial Design
- Micro-Randomized Design
- SMART Design
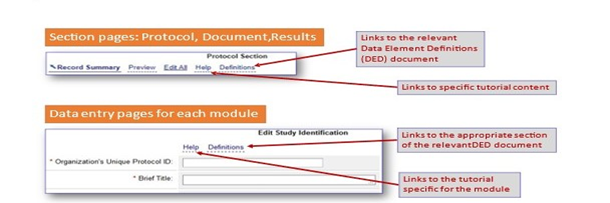
Locating Support and Training Materials in the PRS
Using the Help Menu
The various elements in the “Help” section provide information for protocol data entry, study document submission, and guidance for results submission.
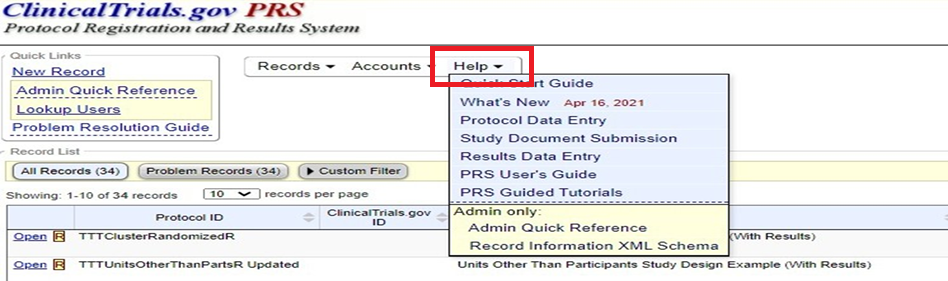
Additional resources for data entry in the protocol section of the application can be found here. It provides information for both interventional and observational studies.
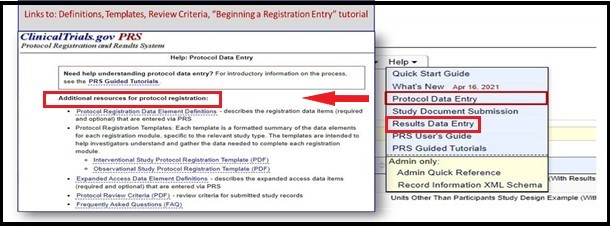
These resources also provide clear guidelines for how to enter results data for various study designs.
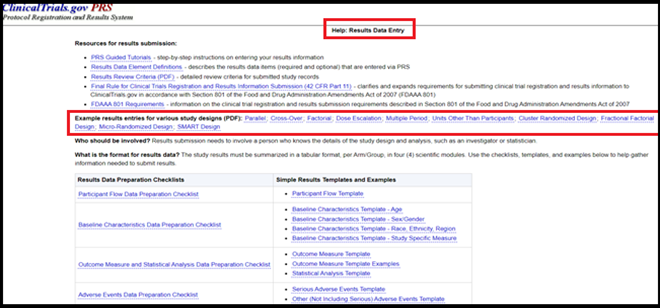
Tutorials are also available to provide more guidance, how to upload required study documents with result submission.
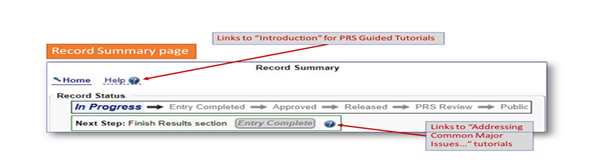
Links on the Record Summary Page
PRS guided tutorials can be found under the “Help” link
Links from HSO Website
The Human Subjects Office (HSO) website provides resources to help researchers with the CT.gov registration process in the ClinicalTrials.gove Investigator’s Guide. If researchers have any questions regarding the registration process, they can contact either the PRS administrator directly or a member of the CT.gov Working Group. This group acts as departmental liaisons to clinical trial investigators. They can help investigators with the registration process and help them understand how to complete different sections of the PRS.

For additional assistance or questions about registering a clinical trial on ClT.gov or using the PRS, please contact us at ct-gov@uiowa.edu or 319.335.6564.
In the News
- The NIH Has the Opportunity to Address Research Funding Disparities, Bill of Health, Petrie-Flom Center at the Harvard School of Law
- People in Areas Hit Hardest by COVID-19 Lack Access to New Treatment Trials, ACRP blog
- Gut Bacteria Change as You Get Older—and May Accelerate Aging, Scientific American
- Listening to a Story Helps Hospitalized Kids Heal, Scientific American
- Preliminary research finds that even mild cases of COVID-19 leave a mark on the brain – but it’s not yet clear how long it lasts, The Conversation

