ClinicalTrials.gov Final Rule Update
By Brian Brotzman

The U.S. Department of Health and Human Services (HHS) has issued a final rule (42 CFR 11) about registering certain clinical trials and providing summary results in a public database for federally and privately supported clinical trials, named ClinicalTrials.gov.
Registration requirements have been in place under FDAAA 801 since December 2007 and researchers have been required to submit summary results since September 2008 and to report certain adverse event information since September 2009, for Applicable Clinical Trials (ACT). The purpose of Food and Drug Administration Amendments Act (FDAAA) 801 was to:
- help patients find a clinical trial for which they may qualify,
- improve the evidence base that informs clinical care,
- increase the efficiency of the drug and device development process,
- improve clinical research practice and build public trust in clinical research, and
- create a platform for information about clinical trials, that would otherwise be unpublished (e.g. non-significant findings).
Despite the implementation of FDAAA 801, information about clinical trials not available from other public sources has continued to be limited, and ClinicalTrials.gov records have not consistently been created or maintained.
This new rule intends to address these concerns. HHS’ goal is to increase public access to information about certain clinical trials of Food and Drug Administration (FDA)-regulated drugs, biological products and devices, as well as improve access to this information to researchers and reviewers. The Final Rule was adopted with this aim in mind, and expands and clarifies the information posted to ClinicalTrials.gov. Updates include reporting on study results for all Applicable Clinical Trials (ACT), severe penalties specified for the investigator and institution for non-compliance, and requiring investigators to indicate if expanded access is available for an investigational new drug (IND). In addition, the National Institutes of Health (NIH) issued a policy with the same effective date that requires registration and posting results for NIH-funded clinical trials.
For more information, please see the HHS news release and NIH Office of Science Policy blog post. [Please note: the current presidential administration has put a temporary halt on this and other new federal regulations, pending review by appointees of the new administration. This halt has not changed the required compliance date of April 18, 2017, and HHS expects all Applicable Clinical Trials (ACT) to register by this date. The Human Subjects Office will continue to provide updates to the University of Iowa (UI) research community as more information becomes available.]
Compliance Dates
The Final Rule, effective January 18, 2017, requires registration and reporting for all Applicable Clinical Trials. Institutions must be fully compliant with this rule by the compliance date of April 18, 2017. A finding of non-compliance could have a significant impact on the funding and the reputations for both the investigator and institution. The FDA has indicated more attention will be given to ClinicalTrials.gov than in the past, and many journals have agreed to uphold these expectations for publication in journals that follow International Committee of Medical Journal Editors (ICMJE) recommendations.
Consequences for Noncompliance
The consequences for noncompliance are severe and affect the entire institution, not just the noncompliant investigator. Institutions found in violation of this rule could be subject to civil penalties of up to $11,383, as well as an additional $11,383 per day, if the issue is not resolved within 30 days. Additionally, federal funding to the institution can be reduced or stopped until the issue is resolved, and this could impact future funding for the institution. Besides civil penalties, responsible parties also face possible criminal penalties for noncompliance with this rule.
Compliance Resources at the University of Iowa
To ensure compliance at the University of Iowa, the Human Subjects Office is developing a compliance program to identify Applicable Clinical Trials, resolving problem records in the Protocol Registration and Results System (PRS), and assist investigators in meeting federal guidelines. PRS administrators are currently reaching out to UI researchers who have clinical trial records in the PRS at ClinicalTrials.gov. The UI has three PRS administrators that manage PRS records for the institution:
- Brian Brotzman (Human Subjects Office; Senior Compliance Specialist)
- Angela Childs (Holden Comprehensive Cancer Center; Protocol Management Associate)
- Cena Jones-Bitterman (Holden Comprehensive Cancer Center; Clinical Trials Research Manager)
These PRS administrators monitor records, and notify record owners and responsible parties of any issues in a record. However, ClinicalTrials.gov has its own PRS reviewers outside of the University of Iowa who review and accept information published on the site. This means the UI record owner (person completing the record), or the responsible party (sponsor-investigator of the study), must respond directly to reviewer feedback from the PRS review team in the PRS system.
In addition to PRS administration, the Institutional Review Board (IRB) review will begin to consider compliance with ClinicalTrials.gov regulations. While ClinicalTrials.gov is a separate entity from the IRB, compliance with federal regulations is something the IRB considers in its review. To facilitate the new expectations of the Final Rule to FDAAA 801, future revisions to HawkIRB and the IRB review process are planned which will assist investigators in determining if their study is an ACT, and in completing the ClinicalTrials.gov record. To further assist the research community, a ClinicalTrials.gov workgroup is being developed to assist investigators and study coordinators with completing study records and entering data in the PRS. Additional materials and trainings to the research community will be disseminated once they become available.
Important Information to Know About Registering in the Protocol Registration and Results System (PRS)
Some important things to keep in mind if you have a study registered in the PRS system are:
- Update records if making any significant changes to the protocol, such as adding a procedure or updating the risks.
- The Principal Investigator (PI) must register a study if it is an Applicable Clinical Trial and if the PI initiated the protocol or holds the Investigational New Drug (IND) application with the Food and Drug Administration (FDA).
- If registration is required, the PI must be named as the responsible party for the record. This is typically indicated in the record by identifying ‘Sponsor-investigator’ as the responsible party type, and selecting the appropriate University of Iowa investigator.
- Registration is optional for studies that are not Applicable Clinical Trials, such as an observational study, which are not subject to the clinical trial reporting requirements. However, if a trial is registered, it must adhere to the expectations of the PRS, such as yearly updates, regardless of whether it is an Applicable Clinical Trial.
- Some medical journals require registration at ClinicalTrials.gov, regardless of whether it is an Applicable Clinical Trial. Investigators are responsible for awareness of journal requirements, and it is recommended that trials be registered prior to enrollment of the first subject to assure publication in a desired journal can be obtained.
- NIH funded projects must register a clinical trial of any phase.
- The PI should use the study HawkIRB number (number only) as the unique protocol ID in the ClinicalTrial.gov record. That means the UI PI must submit the HawkIRB application before registering the study on ClinicalTrials.gov. The PI can add the National Clinical Trial (NCT) number to Section VII.B.1 of the HawkIRB application when it is routed back during the IRB review process.
Resources Available
The PRS provides multiple helpful resources for explaining how to use the PRS and the specific information it is requesting via help menus, user guides (protocol, results), and tutorial videos. For more information, see the HSO website or email ct-gov@uiowa.edu.
HawkIRB Update: Changes in the “Approval” Tab
By Kelly O’Berry

Researchers with outside funding often need additional documentation about IRB approval of HawkIRB forms to share with a study sponsor. In response to your requests, we have enhanced documentation features to generate a ‘printer friendly version’ of the IRB approval materials on the “Approval” tab located on the Project Summary page. Researchers can find this tab in the menu bar across the top of the Project Summary page.
| Summary | Project Details | Attachments | Committee Docs | Research Team | Funding | REFs | Approval | Monitoring |
The Old “Approval” Tab
The HawkIRB system has always kept a record of the approval of all forms submitted for the study. The approval for the New Project form is first and then all subsequent approvals are listed in reverse chronological order. The “Approval” tab already included the following information for each form:
- The type of form – Modification (Mod), Continuing Review (CR) or Modification/Continuing Review (Mod/CR)
- Date approved by the IRB
- The name of the IRB Chair or Chair Designee and date of their electronic signature
- Any administrative codes (references to the federal regulations) that apply to the IRB review of each form
- Link to the IRB approval memo and the date it was attached to the HawkIRB application
- Link to the partial waiver of HIPAA authorization (if applicable)
Other Reviews – This section provides a link to materials from the review of the form by other Human Research Protection Program (HRPP) committees, such as Pharmacy and Therapeutics (P&T), Medical Radiation Protection Committee (MRPC). The researcher can save those documents or print them for the study sponsor. The Human Research Protection Program Flowchart indicates the other committee reviews that are required.
New Content on the “Approval” Tab
There are two new sections in the “Approval” tab: Basic Study Information and Consent Document Updates.
- Basic Study Information – The “Approval” tab now includes basic study information at the top of the page: The IRB number, study title and PI name. This information is helpful when sharing information about IRB approval with the study sponsor. This information is pulled from Section I and II of the HawkIRB application.
- Consent Updates – There is a section on the “Approval” tab with the filename and the approval and expiration dates for the Informed Consent Document(s). The introductory statement indicates whether the approval and expiration dates were updated and if there were any other changes made in the document(s). If a study has multiple Informed Consent Documents, Assent Documents and/or a Record of Consent, those documents will all show up in the “Consent Updates” section.
There is no link to the consent document(s) from this page. Researchers must access the IRB-approved, stamped version(s) of these documents from the “Attachments” tab on the Project Summary page. Researchers who need a record of the changes that were made in the document(s) can go back to the “Form Attachments” page for the HawkIRB form that was just approved to access the last tracked-changes version of the document(s). Click on the “+” to the left of the document attachment name to reveal links to all previous versions of the document since the study was first submitted to the IRB.
Researchers may want to include a date or other indicator of the document version in the document filename(s) so the sponsor can tell that it is a newer version. That may require a change from the standard naming convention the PI and his/her delegates have been using for Informed Consent Documents. Updating the filename is only an option when the consent documents are modified (not an option for Continuing Review-only).
New Feature on the “Approval” Tab
Researchers now have the option to create a ‘printer friendly version’ of the “Approval” tab to share with the study sponsor. This is similar to creating a ‘printer friendly version’ of a draft form (via the link on the right side of the Principal Investigator’s inbox for the draft form) or an IRB-approved project (via the link in the “Project Details” tab). The ‘printer friendly version’ opens in a new window of your browser. It includes the content of the page without the HawkIRB navigation features. A researcher can either print the ‘printer friendly version’ or save it as an html file to share with the sponsor.
Note: There is an approval tab within each individual HawkIRB form that records information about the IRB approval of that form. That tab includes most of the information that is on the main study “Approval” tab. However, the new ‘printer friendly view’ feature is only available in the main “Approval” tab.
We are interested to hear what you think of these new features or if you have recommendations for additional enhancements. Send comments and suggestions to irb@uiowa.edu.
Who’s steering the boat? When PIs (and research team members) “jump ship”
By Becka Simpson
A research study is like a voyage, at some point the boat will dock (the study will close) or the crew will shift (the Principal Investigator (PI) or research team members will leave). This article will explain what to do when the PI leaves the University of Iowa (UI), why addressing this situation is important, and what to do when a research team member leaves the UI or is no longer involved with the study.

The PI is the ‘captain’ of the ship. The PI signs the Assurance document agreeing to take responsibility for the conduct of the research, follow all UI policies and procedures and oversee all study aspects. An engaged, active PI is critical to ensuring the IRB’s trust that the study will be conducted in an ethical manner. The PI must be a UI faculty or staff member or an enrolled student. When the PI leaves the UI or is no longer able to oversee the study, either someone else will take on study oversight as a new PI or the study should be closed. When a PI leaves the University of Iowa, it is best for him/her to engage in active planning for the study’s next steps prior to departure.
Close the Study
The study may be nearing completion. If data collection is complete and the study no longer utilizes identifiable data, it may make sense to close the study. The IRB considers data to be identifiable if there are any ID codes or identifying information (name, date of birth, contact information, etc.) that could be linked back to individual subjects. Working with anonymous or de-identified data is not considered to be human subjects research. IRB approval is not necessary to continue data analysis once the identifiers are removed or if they were never collected or recorded in the first place. So data analysis can continue after the study closes, if the data is not identifiable. However, the study must remain open if linked data is needed so that follow-up can continue or some identifying information must be maintained.
Replace the Principal Investigator
If the study remains open, submit a HawkIRB Modification form to replace the PI with a new ‘Captain’. The new PI will need to generate and sign a new Assurance document, agreeing to oversee the conduct of the study. The PI must obtain all of the other required signatures and attach the document to the HawkIRB application. As with a new study, the IRB will consider whether the PI has the expertise, resources, and support needed to conduct the study.
Unexpected PI Departure
the PI leaves the UI without preparing for his/her departure ahead of time, they’ve left a gap in oversight for the research. The project is left with no one at the ‘helm’ and the IRB will not allow the study to continue in that state. The PI may or may not have retained an “active” UI appointment in these instances. If their University of Iowa appointment has been formally terminated, HawkIRB documents team member de-activations with a red “Yes” in the Deactivated column in Section II.2 of the HawkIRB application, which means that the university HR system or the Registrar’s Office no longer lists that person as having an active affiliation with UI. When HSO staff become aware of a de-activated PI, we send an email notification to the PI, his/her delegates, faculty advisors and the Department Executive Officer (DEO) or Department Chair to alert them to the IRB’s concern about study oversight. The study has five business days to address the lack of an active PI by either closing the study or modifying the study to appoint a new PI. The email notice describes the steps needed to allow the study to maintain IRB approval. If there is no action, the HSO will administratively close the study.
closure is a permanent action. The IRB application cannot be “re-opened” once a study is closed. If the PI wishes to resume study activities after the application is closed, s/he will need to submit a new study application and obtain IRB approval before the research activity may begin again.
De-activated Research Team Members
Research team members can also be “de-activated.” The PI should submit HawkIRB Modification forms, as necessary, to reflect changes in the research team. Team members need to be removed when they are no longer affiliated with the University, as the IRB generally will not provide oversight to them. If a research team member’s expertise was integral to completing study activities or ensuring subject safety, then the PI will need to replace this person with someone else with the same expertise.
Changes in a project’s personnel are to be expected. With a little planning (and notification to the IRB), your study can maintain its course and reach its destination.
External/Commercial IRB Q&A Info Spot: How does the Project Material Obtained (PMO) question influence the HawkIRB review process?
By Kathy Beck

Q: What is the purpose of the Project Material Obtained (PMO) question?
A: This question only applies to HawkIRB applications for commercial IRBs. A sponsor may require a study number for pre-submission negotiation tasks. The Principal Investigator (PI) can submit a partial HawkIRB application by completing sections I through IV, answering “No” in the PMO (“Program Materials Obtained”) section, and attaching the signed assurance document.
The HawkIRB system generates an IRB # when the PI submits an application. Human Subjects Office (HSO) staff will review the partial application and then route it back to the PI in workflow while s/he continues working with the sponsor on the pre-submission tasks.
After the PI has obtained the protocol, consent documents, and applicable materials for Human Research Protection Program (HRPP) committee(s) review, the PI should change the answer to the PMO question to "yes." This will open additional sections of the HawkIRB application. The PI should complete the rest of the HawkIRB application, attach all necessary documents and then route the application back to the IRB through workflow. When the PI submits a complete HawkIRB application, the HawkIRB system will alert all applicable HRPP Committees that their review is required.
Q: Does submitting a partial HawkIRB application speed up HRPP committee review?
A: No. The PI must complete Section V of the HawkIRB application, which opens after PMO answer is “yes,” to trigger a notification to all applicable HRPP committees. The HawkIRB system does not notify the HRPP committees when the response to the PMO question is “no.”
Q: How often do researchers need an IRB number prior to submitting a full HawkIRB application?
A: Most researchers do not need a study number for pre-negotiations; however, the option is available if needed. If you have questions about whether the materials you have are “enough” to submit an application or if you should indicate “no” to the PMO question, please contact the HSO office for guidance (319-335-6564 or uirb-external@uiowa.edu or uiwirb@uiowa.edu). For commercial or external IRB-specific questions, we encourage you to set up a meeting with Kathy Beck (335-7297, kathleen-beck@uiowa.edu) and Anna Mertes, (335-9915, anna-mertes@uiowa.edu).
Herky Hints: Modifications about Funding Source Changes
By Brent Collinsworth

Funding. This 7-letter, 2-syllable word is like magic to a researcher’s ears. After a study is approved by the IRB, it can obtain funding for the first time, receive additional funds from a new funding source, or funding for the study may end. Either way, when funding status changes during the course of a study, you will need to report the change to the IRB by submitting a HawkIRB Modification form. Remember that you always want your HawkIRB application to accurately reflect the details about the study, including the funding source.
When submitting a Modification, there are a couple things to keep in mind. If you add or remove a funding source, make sure that you update the rest of the application and all attached documents to reflect the change. The Informed Consent Document not only lists the funding source but also tells subjects about compensation they will receive for participating in the study. If the funding source or compensation plan changes during the course of the study, make sure the consent document is updated. You can edit the consent document by going to the Attachment Changes tab of the Modification form. Click on the “EDIT” link to the right of the grant application/funding document and then follow the instructions on the page to save the document to your hard drive, make changes, and re-upload it to the application (see example below).
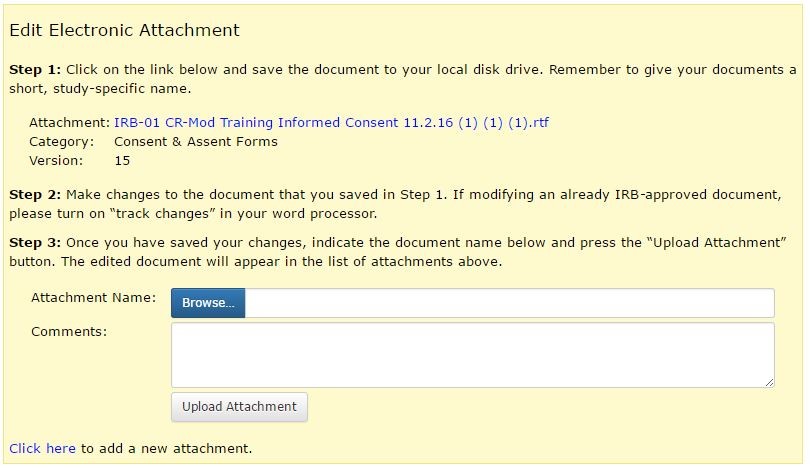
If your project initially had “No Funding” and you add a funding source, make sure to remove the “No Funding” option in Section III. This is not automatically removed when you add new funding. You can remove any entry in Section III by clicking on the “remove” link, to the right of the funding source.
If funding changes during the course of the study, you will need to provide additional information or an explanation to the IRB.
If your funding ends you would remove the funding source from Section III and then explain that to the IRB in Section XIII of the Modification Form, “Other Mod and/or Comments”. Section XIII is an open comment field of the HawkIRB Modification form that allows you to explain changes that you make in the application. Be sure to explain how you will be able to continue the study without funding. If to the study provides compensation to subjects, you will need to explain how you will be able to pay subjects without any funding. If departmental funds will be used for subject compensation, add that funding source in Section III.
you are adding a funding source to an existing IRB application, provide an explanation of how the funding is related to the overall conduct of the study in Section XIII. A copy of the new grant\funding source will likely be required as an attachment on the HawkIRB attachments page. The IRB will conduct a congruency review of the grant and the IRB application.
If your funding ends, you should also remove the attached grant application or funding document from the application. In HawkIRB, all previous versions of an application are archived and still available, so there is no need to keep the grant application in the application. You can remove a document in the Attachment Changes tab of the Modification form. Click on the “EDIT” link to the right of the grant application/funding document and then click “delete” on the far right side of the screen (circled in red, below).

Keep these points in mind so that making changes to the funding source in HawkIRB will be smooth and easy! Most importantly, the HawkIRB application and all attached documents will always accurately reflect the current funding source(s).
IRB Advisor, April 2017: Ethical Issues of Research Recruiting on Social Media
By Brent Collinsworth
The first modern website to meet the current definition of “social media”, Six Degrees, became live in 1997. Since then, social media websites have grown in popularity. In 2017, an astounding 81% of Americans have some kind of social media profile. This explosion in popularity over the last 2 decades has led many researchers to consider utilizing social media as a recruitment strategy. However, there are some potential ethical considerations when recruiting subjects online. Researchers should consider privacy and confidentiality protections to ensure researchers do not compound the privacy risk of someone self-disclosing by publicizing that disclosure further. For instance, if a potential participant states on Twitter that they are feeling severe anxiety, researchers should privately contact them instead of publicly replying to the tweet. This avoids further publicizing their tweet and breaching their privacy. These and other potential ethical concerns (such as investigator transparency) are discussed in an interview between IRB Advisor and Dr. Luke Gelinas, a Harvard Catalyst fellow in clinical research ethics at the Petrie-Flom Center of Harvard Law School. The article is available here. Another article provides a checklist of key points to be aware of when evaluating proposals involving recruitment through social media, available here.
About IRB Advisor
IRB Advisor is a monthly newsletter that contains articles about regulatory issues, informed consent, current events in human subjects protections as well as articles about IRB administrative and management issues. The UI IRB subscribes to this publication as a resource for UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff. Each month the IRB Connection Newsletter features an article from the current issue of IRB Advisor.
The April issue also includes articles about:
- Adverse Event Tracking in Behavioral Studies
- ROMP: The Complex Intersection Between QI and Clinical Research
- IRB Gets New Researchers’ Attention With Visually Clever Infographics
- Here Is a Nutshell Look at Ways to Improve Compliance
- A Consent Consult Helps New Researchers With Informed Consent
- Student Receives IRB Approval to Collect and Display Comments From Sexual Assault Victims
Current and Past Issues
There is a link to current and past issues of IRB Advisor on the Education and Training page of the Human Subjects Office web site. This link provides automatic access to the newsletter from all computers with a University of Iowa IP address.
The University of Iowa username and password cannot be posted on the Human Subjects Office web site. UI researchers may contact the Human Subjects Office to request the username and password to access IRB Advisor from a personal computer. Contact us by e-mail (irb-outreach@uiowa.edu) or call us at 319-335-6564.

