Letter From The Director
New Informed Consent Requirement for Clinical Trials
By Michele Countryman
Welcome back! I hope this spring break provided you rest and relaxation. The Human Subjects Office (HSO) is moving forward with implementing components of the new Common Rule changes. We feel that this particular change will be especially helpful for protecting the rights and welfare of human subjects participating in research.
The Final Revisions to the Common Rule, under section 45 CFR 46.116(a)(5)(i), “requires that key information be included in the beginning of the informed consent in a concise and focused presentation.” To address this requirement, the UI has adopted a consent summary to use in conjunction with the study Informed Consent Document. The consent summary is a one or two page document with fillable sections for the research team to insert information about the study, similar to the current Informed Consent Document template. The expectation is for this document to contain key study related information summarized concisely for the research subject.
The UI adopted these consent summary documents when the National Cancer Institute Cancer IRB (NCI CIRB) option was launched this past July 2016. Three consent summaries are available for use.
- General Clinical Trial Consent Summary
- Phase 2 Clinical Trial Consent Summary
- Phase 3 Clinical Trial Consent Summary
The HSO\IRB will now require the use of a consent summary for all new clinical trial submissions under review by the IRB-01, IRB-03, NCI CIRB, or any other external IRB. The consent summary template can be downloaded from the dropdown options in the Informed Consent category found on the HawkIRB attachments page. While it is not required, we highly recommend that all existing, IRB-approved clinical trials also consider creating and obtaining IRB approval for a consent summary to go along with the Informed Consent Document. The Principal Investigator of an approved clinical trial can submit a modification at any time to make this update.
The intent of the consent summary is to present this key information to the research subject as a supplement to the regular informed consent document. This will provide the subject with an, at a glance, easily understandable, view of what they can expect if they participate in the research project. Informed Consent is not only a document, it’s an ongoing process between researchers and subjects to ensure they are adequately informed and are able to have questions answered as they are considering whether or not to participate in the research. If you have any questions about how to implement this new requirement, please contact the HSO at (319) 335-6564 or irb@uiowa.edu
HawkIRB Carousel Index: What the IRB Asks For and Why
By Brent Collinsworth

If you have generated a draft New Project Application in HawkIRB recently, you may have noticed the new carousel format for the Project Index. The new carousel index will assist researchers with navigating their HawkIRB application and provide background information about each section of the New Project form.
The new index is divided into two sections: the navigation bar on top (1) and the detailed section boxes below (2). The top navigation bar shows the completion status of each section of your application. The detailed section boxes include sub-headings for groups of questions below each section title. You can navigate through the section boxes using the red arrows on either side of the screen, or click on a section number in the top navigation bar to scroll to the section box. You will need to click in the detailed section box to open the application. Finally, there are links to the Attachment page, Other Committee Review Attachments and the Final Submission page between the top navigation bar and the section boxes.

The carousel index differs from the old index format in four distinct ways:
- More information about the topics covered in each section
- Color coding to track completion of the form
- Information about what you need to complete each section
- Information about why the IRB needs the information collected in each section
More Information About Each Section
The previous HawkIRB index format listed the section number and a general title for each section. The sub-headings in the new carousel index indicate the topics covered, including whether multiple topics are covered in a section. For example, Section IV covers the project type and waiver of informed consent. These sub-headings allow researchers to scroll through the index to find where to describe a specific aspect of the study.
The new sub-headings also specify the information covered in of the five parts of Section VII (sections A-E). For example, you can now tell from the index that Section C is about genetic research, Section VII.D is about the recruitment and consent procedures and Section VII.E is about project procedures and compensation.
Color-Coded for Your Convenience
The new index shows you at a glance what sections of the draft HawkIRB application are complete, and what you have yet to complete.
In the top navigation bar on the Index, each section will show up as one of four colors.
- Grey means a section is not required. This is based on current responses in the form. If no answers have been provided, the index will largely be grey.
- White sections are required but not yet started.
- Yellow means at least one or more questions have not been answered in that section.
- Green sections are complete.
The section boxes in the lower part of the index give you more detail about the draft application. Each section and sub-heading under that section can be one of three colors:
- Grey text with no hyperlink indicate sections that are either not required, or questions need to be answered before this section will open
- Blue text indicates the sections that blue are required, but either not started or incomplete. You can click on the blue text to go to that section of the application.
- A green check mark indicates completed section.
“What you need to start”
Another feature of the carousel index is the addition of the “What you need to start” message. The link to these messages are at the top portion of each section of the detailed index box, above the section title. These popup messages list the specific information researchers will need to answer questions in that section of the application. For instance, to complete Section II you will need a list of everyone who meets the regulatory definition of engaged in research, which includes, but is not limited to, having direct contact with research subjects and/or access to their identifiable private information.
The HSO\IRB recommends reviewing all of the “What you need to start” messages to know what you will need to have available prior to starting each section.
“Why we need this information”
The final new feature of the carousel index is the “Why we need this information” message. The links to these messages are at the bottom of each section of the detailed index box. These messages reference the regulations, guidance, and institutional policies that require the IRB to collect this information in each HawkIRB application. For instance, Section III (Funding/Support) discusses five main research-associated organizations that have specific requirements for sponsors and funding: The Office of Human Research Protections (OHRP), Association for the Accreditation of Human Subjects Research Protection Programs (AAHRPP), the University of Iowa IRB, the Department of Defense (DoD), and the National Science Foundation (NSF).
Read the “Why we need this information” to understand the regulatory and policy basis for each section in the HawkIRB application.
Learn More
If you would like more information about the HawkIRB Carousel Index, follow this link to the Carousel handout on the HSO website.
External/Commercial IRB Q&A Info Spot: Processing external/commercial IRB acknowledgements
By Kathy Beck
Q: Do I need to update HawkIRB when the external/commercial IRB of Records sends an ‘acknowledgment’ notice instead of a formal decision from the IRB?

A: The quick answer is “yes”. The UI investigator is responsible for initiating the HawkIRB application, regardless if the UI investigator or the sponsor submits the request to the external/commercial IRB of Record.
Why are we required to complete this step when the University of Iowa is not the IRB of Record? The external/commercial IRBs do not verify the credentialing and training or the compliance with the local institutional policy. Submitting the modification in HawkIRB enables the local context reviews to be completed. HawkIRB is the venue for the University of Iowa Human Research Protection Program (HRPP) committees to complete and document all required reviews.
There are two common scenarios when an external/commercial IRB may provide an ‘acknowledgment’ notice are (1) Investigator Brochure amendments and (2) revisions to research team members. This article will provide an overview of the HawkIRB modification submission for a study utilizing an external/commercial IRB, including how to document the external/commercial IRB’s acknowledgement in HawkIRB.
Basic process:
Whenever there is a change in the study design, procedures, materials or research team, the Principal Investigator (PI) must submit the change to the external/commercial IRB and to the University of Iowa (UI) IRB concurrently. All submissions to the external/commercial IRB of Record require a submission in HawkIRB.
Submit a HawkIRB Modification form to the UI IRB
For Investigator Brochure amendments, submit this Modification form after you have submitted the change to the external/commercial IRB. For research team member changes, submit the Modification form before you submit the change to the external/commercial IRB (see below for additional instructions for changes in team members).
- Communication with the external/commercial IRB - Use the ‘Edit’ function in HawkIRB to attach the form (document or e-mail) submitted to the external/commercial IRB. This will save the new version on top of the previous document attached in the attachment category for ‘Copy of external IRB application.’
- Revised documents - Attach documents submitted to the external/commercial IRB for consideration.
- Changes in HawkIRB - Revise the HawkIRB application to reflect the changes. For Investigator Brochure amendments you will need to change Section V. For research team member changes you will need to change Section II. But you might need to revise other sections of the HawkIRB application. Be sure to make changes consistently throughout the application form and attached documents.
When the external/commercial IRB ‘acknowledges’ the submission, the HawkIRB Modification form will either be in the PIs inbox or with the IRB.
- If HawkIRB application is in PI’s inbox: Use the ‘Edit’ function to attach a copy of the ‘acknowledgment’ in the attachment category for, ‘Copy of external IRB application’. Then you may return the form to the IRB for processing
- If HawkIRB is with the IRB: Contact the HSO (see contact information below) to request that the application be routed back to PI. Then follow the instructions above for attaching the acknowledgment and returning the form to the IRB.
Here is how to contact the HSO:
- Phone: 335-6564
- Email for studies using WIRB as IRB of Record: uiwirb@uiowa.edu
- Email for studies using other external/commercial IRB of Record: uirb-external@uiowa.edu
Additional Information: Research Team Changes
If the only change to the study is a revision to the research team member(s), submit the HawkIRB Modification form to add or remove a research team member before submitting the change to the external/commercial IRB. That ensures that new team members have completed the CITI training for human subjects protection and the conflict of interest disclosure. The University of Iowa is responsible for the verification of the credentialing and training regardless of the IRB of Record.
There is a different process for changes in the PI. The external/commercial IRB will issue a formal approval memo rather than an acknowledgment. PI changes will be addressed in a future External/Commercial IRB Q&A Info Spot.
Herky Hint: Breaking the News to the IRB – ONLY IF the Subject or the Subject’s Partner Gets Pregnant
By Terese Barenz

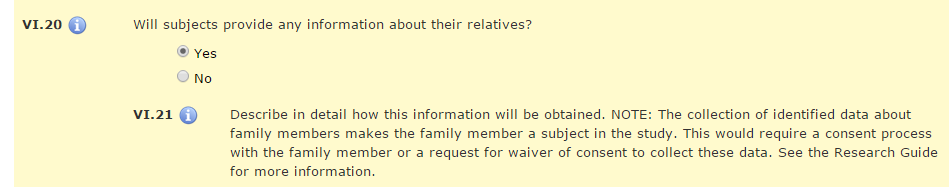
Some studies plan up front to collect information about other people (usually relatives or family members) from the enrolled subjects. For other studies, this type of data collection is contingent on a certain condition, such as pregnancy in the subject or their partner. For example, in studies involving investigational products that allow the enrollment of women of childbearing potential and/or males who are willing to use effective methods of contraception, the researcher may need to collect additional data if pregnancy occurs. So how does the research team apprise the IRB that it will only collect pertinent pregnancy information when the subject or the subject’s partner gets pregnant?
Researchers should communicate plans for this type of data collection in the New Project Application. HawkIRB Section VI.20 asks, “Will subjects provide any information about their relatives?” If male subjects will be asked to provide follow-up information about the outcome of the partner’s pregnancy or female subjects will be asked to provide information about the outcome of their pregnancy, the correct response in this section is “Yes.” The details about how the information will be collected and what information will be obtained should be provided in the following two questions. The IRB only needs to review and approve these plans if a subject/partner becomes pregnant. In the New Project application, the Principal Investigator can indicate plans to submit a HawkIRB Modification form if this additional data collection becomes necessary.
Since the information will be collected only if pregnancy occurs, an appropriate response here is:
If a subject or subject’s partner becomes pregnant, we will submit a modification form to explain how we will obtain the information to follow the pregnancy to completion.

Once again, the response should explain that a modification form will be submitted. An appropriate response would be:
If pregnancy occurs, we will submit a modification form to describe the data that will be collected and/or specify the name of the data collection forms attached in the “Relative/Proxy Data Collection Instrument” category on the attachment page (i.e., sponsor’s Pregnancy Report Forms).
If a pregnancy does occur during the course of the study, then the research team will submit a modification form at that time, providing the specific plans for data collection in Section VI.21 and VI.22. This may require submitting a new or revised protocol or a document outlining the sponsor reporting requirements. The IRB also has additional responsibilities when vulnerable populations are included in a study. Therefore, they will review this new information to determine if the study still satisfies the criteria for IRB approval as well as evaluating whether the study complies with the “Additional Protections for Pregnant Women, Human Fetuses and Neonates Involved in Research” as outlined in 45 CFR 46 Subpart B. When the IRB makes these determinations, they are documented on the IRB approval page for the modification.
If a pregnancy does not occur in a subject/subject’s partner, then a modification to these sections will not be necessary.
Separate HawkIRB applications MAY BE REQUIRED for research at the VA Hospital and the University of Iowa. How do I know?
By Tony

As of February 15th 2016, research conducted at both the Iowa City Veterans Administration Health Care System (VAHCS) and the University of Iowa can no longer be described in the same HawkIRB application. The IRB-03 board only reviews research occurring at the Iowa City VAHCS while the IRB-01 and IRB-02 boards only review research occurring at the University of Iowa. Researchers at both the VAHCS and University of Iowa will need to assess where activities will take place and what resources will be used for their proposed study to determine if one or two HawkIRB applications are required.
What does this mean for research projects conducted at both locations?
Many Principal Investigators have already ‘split’ their existing projects into separate HawkIRB IRB-03 and IRB-01/02 applications when their project involves activities at both the VAHCS and the University of Iowa. This transition is expected to be made on or before the time your study’s next continuing review form is due. Also, many IRB-03 existing projects involving research at the VAHCS only have converted the IRB of record in their HawkIRB application to ‘IRB-03 only’ (question I.1 in the application).
If want to submit a new research application for IRB review where you are uncertain if the VAHCS is involved, there are two main things to consider when determining which IRB will review your application:
- Where are the research activities occurring and
- What resources are being used for your research.
This will determine whether your project will require two separate IRB applications.
Where are the research activities occurring?
If all the research activities (access to medical records, research procedures, storage of research data/specimens, etc.) occur in one institution/location, either the VA or University of Iowa, you will only submit one IRB application. Studies where all research activities occur at the VA will submit an IRB-03 application and select ‘IRB-03 only’ for question I.1 in the HawkIRB application. Studies where all research activities occur at the University of Iowa will select “IRB-01” for biomedical research or “IRB-02” for social/behavioral research in question I.1 in the HawkIRB application. If your project clearly involves research activities at both institutions/locations, you will be required to submit separate HawkIRB applications. One application to IRB-03 describing only the research occurring at the Iowa City VAHCS and the second application submitted to the appropriate IRB-01 or IRB-02 board describing only research activities occurring at the University of Iowa.
For research occurring at both the UI and the VAHCS, one location either the University of Iowa or the Iowa City VAHCS, will need to be identified as the “lead coordinating center” while the other location will be identified as a “participating site” in section VII.A of the IRB application. VII.A.2 asks whether the “project is also being conducted by other researchers at their own sites (e.g. a multi-site collaborative project).” This question should be answered “yes” to allow additional questions to gather information regarding the collaborative relationship.
What resources are being used for my research?
Some studies will have their primary research activities at one location but use resources from the other institution/location. These resources may include:
- Medical records
- Specimen storage
- Data Storage
- Use of office space/computers
- Funding sources - Is funding issued from UI Division of Sponsored Programs (DSP) or the VA Research Foundation\Research Office?
- Research team members and/or salary support - This is determined based on their appointment location and percent effort allocations. Zero dollar VAHCS appointments should be carefully considered. If the individual is a 100% compensated UI employee, activities are considered UI efforts and require an IRB-01 application.
- Manuscript preparation
- Data analysis
Projects that have their primary activities at one institution/location but also include any of these resources or activities at the other institution/location will be required to submit separate HawkIRB applications for each location. Investigators will submit separate HawkIRB applications for IRB-03 and either IRB-01 or IRB-02 review. In Section I.4, each HawkIRB application should include a reference to the other application submitted for review by a different IRB. Once the applications have been submitted to the IRB, these references can include the HawkIRB number for the other application.
This is a brief overview of what researchers should consider when submitting new projects for IRB approval. For more detailed information, review the Weblink found under Research at the VAHCS on the HSO website.
For questions or guidance on existing IRB-03 studies or how to proceed with a new IRB-03 application, please call (335-9848) or contact me by email (tony-quinlan@uiowa.edu).
Top Monitoring Findings of 2016 – And How to Prevent Them!
By Patricia Katopol, PhD

IRB Compliance and Education staff know that monitoring can be stressful for Principal Investigators (PIs) and research team members. Even after working diligently on the HawkIRB application, the monitoring visit can identify responses that are incomplete or not as clear as they could be, resulting in a finding and required action to modify the information. To illustrate the most common problems in HawkIRB applications, the Human Subjects Office proudly presents Top Monitoring Findings of 2016 – and how to prevent them in 2017:
Runners Up:
- Section III.1 Funding Source –Information about funding is part of the initial HawkIRB application. However, the funding source can change during the course of a study and it is a problem if this is not reflected in the HawkIRB application. A HawkIRB modification form is required if new funding is awarded or if a funding source ends. “All original sources of funds that will be used to support the study must be listed in Section III of the HawkIRB application. This includes “pass-through” funding, in which the Principal Investigator of the study and identified in the HawkIRB application, is not the Principal Investigator of the funding grant.” (UI IRB Standard Operating Procedures (SOP) & Researcher Guide Section II, Part 4. HawkIRB Section III Funding/Support)
- Section VII.D.1 recruitment method and material list – IRB Compliance and Education staff found inaccurate or incomplete descriptions of study recruitment methods and materials. All recruitment methods must be listed in the application and approved by the IRB. This includes the text for ads, posters, and mass emails. Changes to recruiting methods, such as revising the language in an advertisement or removing a brochure, require modification and IRB approval. “Only the currently approved recruitment materials and methods should be used to recruit subjects.” (UI IRB Standard Operating Procedures (SOP) & Researcher Guide, Section II, 11.A.ii Recruitment materials & methods)
- Section VII.E.6 study procedures – During the monitoring visit, the Compliance and Education Specialist may identify the need for a better description of study procedures or find that the procedures have changed from those approved by the IRB. For example, if you work with an off-campus lab, provide the lab’s name and location/organization. As with recruitment methods and materials, the PI must submit the new description to the IRB as a modification.
- Sections VII.D.5 and VII.D.6 confidentiality protections for protected health information (PHI) used for recruitment purposes – Applications can be too general in descriptions of how health identifiers will be protected, so that the monitoring visit becomes an occasion to review the requirements for a partial waiver of HIPAA authorization to use PHI to identify potential subjects.
Best practice for responding to the question in Section VII.D.5 includes providing the role or status (such as ‘research team member’) of persons involved in obtaining and transporting specimens, the location of the data, and who has access to that location. Stating that ‘data will be kept in a secure location’ is insufficient. Think about who is doing what, where, and how in regards to data collected from medical records specifically for recruitment purposes. Be clear in your response if the data will remain in an electronic system such as EPIC, and will not be printed or stored elsewhere. (HawkIRB Help Message VII.D.5).
Section VII.D.6 requests information on destroying identifiers to protect PHI. Remember to provide information about how you will remove the identifiers of people who do not enroll in the study as soon as possible consistent with the conduct of the study – this may include waiting until enrollment is complete to avoid re-contacting people who have decline to participate. See Partial Waiver of HIPAA Authorization.
Third Place
- Section VII.D.29 enrollment process for adult subjects –The researcher is asked to address three specific aspects of the study in Section VII.D.29: (1) the recruitment process, (2) the enrollment process, and (3) how the researcher will minimize coercion or undue influence in the consent process. Recruitment refers to sharing information about the study with potential subjects. This response should describe the use of all recruitment methods listed in Section VII.D.1. The enrollment process includes sharing detailed information about the study procedures, risks, benefits, etc., allowing subjects to ask questions and think about participation, as well as actually signing the consent document. Minimizing coercion or undue influence requires, among other things, that the investigator emphasize that medical care will not be affected by refusal to participate in the study, that participation is voluntary, and that the potential subject may ask questions about the study. “The Principal Investigator must accurately and completely describe the study procedures in the HawkIRB application.” (UI IRB Standard Operating procedures (SOP) & Researcher Guide: Section II, 1.A Beginning the IRB New Project Application)
Another frequent finding is that researchers do not include information on their process for reconsenting minors who become adults during the study – such as by mailing out a new consent, consenting in person, or faxing a consent form. They may also fail to describe how they will ensure that a subject is capable of providing consent - the PI may find Evaluation to Sign an Informed Consent Document for Research to be helpful for this. Detailing the process when consents are mailed out is also a common finding. For example, the PI must describe procedures if returned consents are incomplete or incorrectly completed. Recognizing the problems with mailed-out consent forms and details about how the study will address them should be in the application. (UI IRB Standard Operating Procedures (SOP) & Researcher Guide, Section II, 17.A.ii Minors aging up process (Minors Reaching the Legal Age of Consent While Enrolled in a Study Policy))
Second Place
- Section II.2 research team list – Investigators usually remember to submit a Modification in HawkIRB to add new team members, but often forget to update the research team roster when a team member leaves the UI or is no longer involved with the study. Adding or removing team members affiliated with the University of Iowa is an easy modification that can be approved by the IRB within 24-48 hours. “The HawkIRB application should list all individuals who have contact or interactions with research subjects or their private, identifiable information as research team members.” (UI IRB Standard Operating Procedures (SOP) & Researcher Guide, Section II, Part 3: HawkIRB Section II Research Team, 3.8 UI Team Members).
And the winner is….
- Section X.4 Information collected list – This section requests information about the collection and storage of paper and hard copy records, electronic records, and biologic samples. Often, the response does not have the level of detail necessary for a full picture of confidentiality protections for all study data. “The description should include the storage location, who has access to the data and/or specimens, and the individual responsible for maintaining IT security (typically the investigator’s departmental IT person), as well as the person responsible for the storage of specimens.” (UI IRB Standard Operating Procedures (SOP) & Researcher Guide, Section II, Part 15: Section X Privacy & Confidentiality, 15.8 Data and/or Specimen protections)
Researchers can take steps to improve the quality of HawkIRB applications and reduce the number of monitoring findings, such as having several team members read through all HawkIRB applications before submission. Of course, when in doubt, please contact the HSO at 319.335.6564 or irb@uiowa.edu for assistance. We are happy to work with you.
IRB Advisor, March 2017: The New Common Rule- Changes, Challenges, and Central IRBs
By Brent Collinsworth
The March IRB Advisor has two separate articles about the revisions to the Common Rule, 45 CFR 46, Subpart A (Common Rule Change Took Six Years to Complete — And Could Be Upended in 30 Seconds & Final Common Rule Is An Improvement, But Leaves Some Questions Unanswered). We are spotlighting both of these articles this month.
The Code of Federal Regulations for the Protection of Human Subjects, published in 1991 by the Department of Health and Human Services’ Office for Human Research Protections (OHRP), is the federal policy overseeing all federally funded human subjects research. It has stood largely unchanged since its original publication date. In the last decade, many researchers and policymakers have encouraged OHRP to release an update to the common rule to modernize regulations, enhance protections to subjects, and reduce burden and ambiguity to researchers. Efforts to answer this call began:
- In 2011 with the Advanced Notice of Proposed Rulemaking (ANPRM).
- Next, the Notice of Proposed Rulemaking (NPRM) released in 2015 after comments on the ANRPM were taken into consideration.
- Finally, the Final Rule released and published in the Federal Register on January 19, 2017.
It has been a long and winding road to get to the new Common Rule but the final outcome of these revised regulations is still uncertain. The Common Rule revisions may be overturned through the Congressional Review Act, a law that gives Congress the prerogative to review and overrule all federal regulations passed in the last 60 legislative days of the previous president’s term. President Trump’s recent executive order requiring a removal of two regulations for every one regulation issued also leaves the Final Rule vulnerable to nullification. IRB officials and experts are cautiously optimistic, however. The new common rule appears to reduce regulatory burden on researchers and administrative burdens on IRBs, which is both helpful and consistent with the President and congressional Republicans’ plans to reduce federal regulatory burden.
Until the implementation of the Final Rule is certain, IRBs across the country (including our own) are on hold to make the policy and procedural changes necessary to comply with the Common Rule revisions. These IRB Advisor articles describe some of the changes in the revised Final Rule:
- Removing pregnant women as a class of vulnerable subjects
- Changing the categories of research that qualify for expedited review and exempt status
- Most studies that qualify for expedited review will no longer need continuing review by the IRB
- Strongly emphasizing on data protection and privacy
- Removing the Federalwide Assurance “checkbox”
- Mandating institutions to use a single IRB for multisite studies
The second article focuses on the implications of the last two points.
All institutions receiving federal research funding support have a Federalwide Assurance, or FWA, with the Department of Health and Human Services. This assurance documents the institution’s commitment to follow all regulations in 45 CFR 46 and ensure that all federally funded research is conducted in compliance with the regulations. In the past, the FWA had an option for the institution to voluntarily extend their FWA to cover all human subjects research, regardless of funding source. This voluntary agreement was indicated by selecting the option under the “applicability” section ofthe FWA. The Final Rule removed that option, giving institutions more flexibility in the review of non-federally funded research.
One of the other proposed changes in the Final Rule is the requirement to use a single IRB for multisite research. When this mandate was first introduced in the Notice of Proposed Rulemaking (NPRM), it received over 300 comments from the public. The single IRB requirement in the Final Rule added some flexibility in implementation. Institutions must comply with the mandate, but now have the ability to conduct additional institutional reviews on these projects. Further explanations about this requirement will be necessary, via guidance documents and templates from OHRP.
About IRB Advisor
IRB Advisor is a monthly newsletter that contains articles about regulatory issues, informed consent, current events in human subjects protections as well as articles about IRB administrative and management issues. The UI IRB subscribes to this publication as a resource for UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff. Each month the IRB Connection Newsletter features an article from the current issue of IRB Advisor.
The March issue also includes articles about:
- Strategies to Better Manage Noncompliance
- Inexact Science: The Complicated Quest To Replicate Research
- Clinical Trials: More is Not Necessarily Better
- Military IRBs May Err on the Side of Bureaucracy
- Teleconsent Boosts Recruitment of Rural Research Participants
Current and Past Issues
There is a link to current and past issues of IRB Advisor on the Education and Training page of the Human Subjects Office web site. This link provides automatic access to the newsletter from all computers with a University of Iowa IP address.
The University of Iowa username and password cannot be posted on the Human Subjects Office web site. UI researchers may contact the Human Subjects Office to request the username and password to access IRB Advisor from a personal computer. Contact us by e-mail (irb-outreach@uiowa.edu) or call us at 319-335-6564.
Continuing Education Credits
Individual newsletter subscribers can receive 1.5 AMA PRA Category 1 Credits™ or 1.5 nursing contact hours for reading an issue and completing an online test. However, since this is an institution-level subscription, UI researchers must purchase access to an individual issue for $40, or purchase a full subscription, to receive CME/CE credits. Visit the AHC Media web site for information about subscription options.

