Index
HSO/IRB Return-to-Campus Notification
Incomplete HawkIRB Applications Will Now be Returned Without Review
Individual Research Results: To Share or Not to Share
HawkIRB Automated Feature: Creating a ClinicalTrials.gov record (Push to PRS)
IRB Advisor Newsletter, May and June 2021
In the News
HSO/IRB Return-to-Campus Notification
The Human Subjects Office will resume in-person compliance monitoring visits beginning August 2021. We will change to a hybrid model for HawkIRB trainings, IRB presentations and Office Hours in September 2021, to allow attendance in person or virtually.
Look for detailed information regarding Fall 2021 education sessions and Office Hours on the Education and Training page by mid-August and in email announcements after the beginning of the semester. We look forward to “seeing” you this fall! Send questions to irb@uiowa.edu or call the Human Subjects Office at (319) 335-6564.
Incomplete HawkIRB Applications Will Now be Returned Without Review
By Kelly O'Berry, BS, CIP
While those responsible for approving HawkIRB applications work hard to turn them around as quickly as feasible, there are some actions researchers can take to improve turnaround time. Beginning in the Fall 2021 academic semester, there are five reasons the HSO/IRB will return an application without review.
In order to perform a thorough and timely review of research protocols, the IRB must have complete, accurate, and understandable information about the proposed study. Applications that lack sufficient detail to address regulatory requirements, are missing attachments, or whose language is too technical for reviewers of various backgrounds to understand, require disproportionate IRB resources and slow down the review of all projects. Submitting complete, accurate, and compliant applications through HawkIRB helps streamline the IRB review process for everyone.
Here are five reasons HawkIRB applications will be returned without review:
- They do not meet federal requirements for protocol approval. HawkIRB forms prompt the PI to provide information so the IRB can verify that the project satisfies the federal regulatory criteria for IRB approval (45 CFR 46.111 and 21 CFR 56.111). As the expert on the study design and procedures, it is the PI’s responsibility to provide a detailed, plain-language description of the study in the HawkIRB application, even if they assign the task of preparing the form to a HawkIRB Delegate. It is essential that the PI provides the information requested in each question of the HawkIRB application so the IRB can review the form.
- The descriptions of the study design and/or the study procedures are incomplete. Be sure to include details about all aspects of the study procedures, including the recruitment and consent processes and confidentiality protections.
- Only partial answers are provided in response to IRB questions. Be sure to read each question carefully and provide the information that is requested. Also, address both parts of a two-part question.
- There are multiple inconsistencies in the description of the study. These may be inconsistencies between responses in the application or between the application and the attached documents.
- Missing, incorrect or incomplete attachments. This includes incorrect or missing signatures on the Assurance Document, attaching a template document rather than a study-specific consent document, or just missing documents related to the conduct of the study
PIs should make sure their HawkIRB Delegate(s) are fully trained to use the HawkIRB system and are knowledgeable about their role preparing forms on behalf of the PI. The HSO/IRB strongly encourages PIs to use the resources available within their college or department for assistance.
HSO/IRB has developed a number of educational resources, summarized on the HSO website, to help researchers prepare complete HawkIRB forms. These resources include:
| Educational Resource | Description |
|---|---|
| HawkIRB Training Sessions | There is a 4-part HawkIRB Training series to help PIs and their Delegates learn how to fill out the New Project form (Parts 1 and 2), forms you need to submit after IRB approval (Part 3), and forms for the single IRB (sIRB) model) (Part 4). This training is highly recommended for first time users of HawkIRB. |
| Recordings of HawkIRB Trainings | If you are not able to attend a live HawkIRB training session, there are full recordings and section-by-section recordings in the IRB ICON Course for Researchers. The portal to this ICON Course is only available on the Education & Training page of the Human Subjects Office website. |
| HawkIRB Carousel Index | At the top of each white box in the HawkIRB New Project form Application Index, there is a link to information about “What you need to start” to complete that section of the form. The PI or HawkIRB Delegate needs all of this information to fully respond to each question in this section. At the bottom of each white box is a link to “Why we need this information.” This includes references to federal regulations and institutional policies. |
| HawkIRB Help Messages | Look for the little blue circles with “i” for “information” (  ) associated with most of the questions in HawkIRB application forms. These Help messages provide guidance to help researchers include all of the necessary information requested in the question. ) associated with most of the questions in HawkIRB application forms. These Help messages provide guidance to help researchers include all of the necessary information requested in the question. |
| Printer Friendly View | This feature in HawkIRB allows researchers to review the entire draft form prior to submission. You cannot edit the form in this view, but you can identify discrepancies or insufficient responses. In the PI’s HawkIRB inbox, in the “Draft Forms” section, click on the “review” link to the right of the project title. You can read each section from this page OR click the “View a printer friendly version of this form” link. This opens in a new tab in your browser and you can save it as an html file. |
| Educational Tools | There are Educational Tools about topics that might be useful for researchers to know about as they design and conduct their research, such as Alternatives to In Person Informed Consent, Data Security, Exempt Categories of Research, and other topics. |
| Experienced Study Coordinators or Principal Investigators | Most colleges/departments have one or more persons who have experience with preparing HawkIRB forms. People who are new to using the HawkIRB system are strongly encouraged to use the resources available within their college or department. |
| Faculty Advisor (FA) of a Student Principal Investigator | Student PIs are required to name their FA as their HawkIRB Delegate. The expectation of the IRB is that the FA will assist the student PI in developing the study design and preview the draft HawkIRB form prior to submission to the IRB. FAs are expected to ensure the student PI’s draft HawkIRB submission is complete and ready for review. Many student PIs are first-time users of HawkIRB and active involvement by the FA can prevent issues that slow down the IRB review process. |
| Human Subjects Office (HSO) Staff | HSO Staff are a great resource when researchers have questions about how to fill out HawkIRB forms. You can attend IRB Office Hours, via Zoom during the pandemic, (no appointment necessary) or call the Human Subjects Office at 319-335-6564 for assistance before you submit the HawkIRB form. |
| ICTS Research Support | The Institute for Clinical and Translational Science (ICTS) offers Research Study Coordinator support and regulatory services for study teams that do not have dedicated research support staff. |
[NOTE: For more information about the importance of submitting HawkIRB applications that are ready for review by all Human Research Protection Program (HRPP) committees, review the slides and recordings from the “Getting on the Same Page” lecture series that was presented to the research community in February 2021. Slides and recordings are posted in the IRB ICON Course for Researchers.]
Individual Research Results: To Share or Not to Share
By Kelly O'Berry, BS, CIP
Biomedical research studies can involve a variety of tests from blood tests to imaging to genetic testing. But there are different perspectives, approaches and restrictions regarding whether to return individual results. This article will explore the issues related to sharing individual research results and provide guidance for UI researchers.
Definitions and Scope

The term “individual research results” refers to findings that have potential health or reproductive importance for the individual subject that is discovered during the course of the study and is related to the study aims (Wolf et al., Genetics in Medicine, 2012, p. 364). An example would be a breast cancer prevention study with results of a CT scan that indicates the subject may have breast cancer. These findings could occur at baseline (beginning of the study), during the study, or at the end of the study (after data analysis).
This term should not be confused with “incidental findings”, which are findings beyond the aims of the study. In the previous example, an incidental finding could be a CT scan that indicates the possibility of lung cancer in a study about breast cancer.
This article will focus on the return of individual research results. It will not address the dissemination of research data and findings, as required by Section 801 of the Food and Drug Administration Act (FDAAA 801) on ClinicalTrials.gov. For more information about this topic, see the ClincalTrials.gov Requirements page of the Human Subjects Office website.
Sources of Information and Guidance
This article is based on three significant sources of information regarding the return of individual research results. The first are the 2016 Secretary’s Advisory Committee on Human Research Protections (SACHRP) recommendations to the Secretary of the U.S. Department of Health and Human Services (HHS) regarding the return of individual research results, including incidental findings. SACHRP recommends returning individual research results, not only for clinical research but also for genomics research and social/behavioral research. SACHRP felt that the arguments for returning individual research results was strongest for clinically relevant results.
Secondly, a 2018 report from the National Academies of Science, Engineering and Medicine (NASEM) provided guidelines for individual research results from biospecimen testing. This report includes recommendations for stakeholders (including investigators, sponsors, research institutions and IRBs) that will help to accomplish the following:
- Support decision making regarding the return of individual research results on a study-by-study basis
- Promote high-quality individual research results
- Foster participant understanding of individual research results
- Revise and harmonize current regulations
Finally, the revised Common Rule (45 CFR 46), published in January 2017 and took effect in January 2019, includes a new required element of consent to indicate whether the researcher will disclose clinically relevant research results, including individual research results, and under what conditions they will be disclosed. Researchers must address this in the Informed Consent Document and during the consent process.
The Food and Drug Administration (FDA) regulations (21 CFR 50 and 56) are silent regarding the return of individual research results, although it is anticipated that the FDA regulations will eventually be harmonized with the revised Common Rule (45 CFR 46). However, the FDA’s ICH E6 Good Clinical Practice guidance states “The investigator/institution should inform a subject when medical care is needed for intercurrent illness(es) of which the investigator becomes aware.” Additionally, the FDA guidance document Investigator Responsibilities – Protecting the Rights, Safety and Welfare of Study Subjects says the investigator should inform subjects when medical care is needed.
Exploring the Issues
According to the NASEM report, the tension about whether to provide individual research results comes from “a conflict in core values – the desire to respect the interests and desires of research participants by communicating results contrasted by the responsibility to protect participants from uncertain, perhaps poorly validated information.” Researchers must decide not only whether to share results of tests related to the research, but also incidental findings that are not the focus of the study but might have significant health implications for the individual subject. The researcher must consider how important they are, whether they are well verified, and if they are even actionable. Appendix D of the NASEM report include a white paper on perspectives and underpinnings of the return of individual-specific research results (Morreim, p. 399).
In the past, it was uncommon for researchers to provide individual research results. The Informed Consent Document either did not mention it at all or explicitly stated that the researcher would not provide the individual results. There were several reasons for this:
- Concern about return of inaccurate results
- Risks associated with the misinterpretation of results
- The perspective that research is for the benefit of society, not the individual subject
- Concern about therapeutic misconception – a misunderstanding about the distinction between research and clinical care
- Restriction on returning results from laboratories that were not certified by the Centers for Medicare and Medicaid Services (CMS) according to the Clinical Laboratory Improvement Amendments (CLIA)
However, many research subjects expect to receive individual test results; even when the Informed Consent Document clearly states that these results will not be provided.
The ethical foundation for the return of research results is found in the ethical principles of Respect for Persons and Beneficence, as described in the Belmont Report.
Respect for Persons
Respect for Persons requires researchers to provide sufficient information, in a language the subject can understand, so that they make an informed decision about whether or not to participate. Voluntary participation is key to the concept of Respect for Persons. According to the SACHRP recommendations, one way to recognize the contribution of research participants is to share individual research results. The information could prove valuable to the subjects themselves or their families. These do not need to be clinically relevant results. It may be reason enough to share results that would be of interest to subjects. However, SACHRP acknowledged that research can still be conducted in a respectful manner even without providing individual research results. People may still be willing to participate to contribute to the advancement of science.
Beneficence
The ethical principle of Beneficence requires researchers to maximize potential benefits to subjects while minimizing the potential risks. It’s possible that returning individual results could lead to harm or discomfort (physical, psychological, financial, reputational or social), and it may not be appropriate to do so. SACHRP recommends that sponsors and researchers consider whether to return individual results during the study design stage.
Benefits and Risks to Sharing Individual Results
There are some potential benefits to returning results. Sometimes sharing individual research results is required as a condition of funding (such as with community based participatory research). Sharing these results may increase transparency and trust between the researcher and subjects and make it easier to recruit and retain subjects if they receive individual-level results. And in some cases, there may be a moral imperative to share individual research results or incidental findings.
But the risks remain that subjects could make health decisions based on inaccurate or misinterpreted information. There could be adverse psychosocial effects of knowing about risk of disease for which there is no treatment or other circumstances that could be upsetting to subjects. It can require additional time, personnel, and resources to provide these results, which could exceed the limits of the study budget. There are legal liabilities for the institution as well, whether or not the researcher returns individual research results.
Just as researchers have a variety of reasons not to provide individual research results, subjects have a variety of reasons for deciding not to receive them. They may prefer to “not know” about something to do with their health, or they may want to protect themselves or others. Either way, researchers must disclose the plans for sharing test results in the Informed Consent Document and during the consent process. If results are provided, subjects should be given the choice about whether to receive them.
UI IRB Guidance
The UI IRB Standard Operating Procedures and Researcher Guide does not include any specific guidance about return of individual research results beyond the regulatory requirement for disclosure in the Informed Consent Document. However, there is guidance about allowing parents to decide whether to receive genetic analysis for children enrolled in research (Section II, Part 10). Upon reaching the age of majority, if the research is ongoing, the now-adult subject may request disclosure of the results of genetic testing.
Also related to research involving genetic testing, the UI IRB SOP and Researcher Guide includes a section on incidental findings (Section II, Part 10.C.i) that provides similar guidance about the option for parents to choose whether to receive these results and the opportunity for the subject to request results when they reach the age of majority.
To address the additional regulatory requirements of informed consent, researchers must make decisions about returning individual research results based on the type of results, whether they were received from a CLIA certified lab, if the research team has staff who are qualified to read the results, the usefulness and trustworthiness of the results, and any standards or guidance within their discipline. The IRB will review and evaluate plans proposed by the researcher based on the applicable Criteria for IRB Approval (45 CFR 46.111 and 21 CFR 56.111). If the researcher modifies the study to add the sharing of individual research results, the IRB will need to consider whether the researcher must re-consent previously enrolled subjects.
HawkIRB Application
Study procedures
The HawkIRB application does not have specific questions about plans to return individual research results. However, this is considered a study procedure, so researchers should describe this in Section VII.E.6 (Provide a detailed description in sequential order of the study procedures following the consent process). The HawkIRB application should include how the researcher will provide the results, when this will happen, and who will provide them. Typically, the researcher will refer the subject to their own healthcare provider for additional testing and diagnosis. The researcher should plan for what to do if a subject does not have a healthcare provider or cannot afford to seek further care from a healthcare provider.
Risks
Researchers should describe any risks associated with the return of individual results in Section VIII.1 (What are the risks to subjects, including emotional or psychological, financial, legal or social and physical?). This could include risks from receiving or not receiving the results.
Informed Consent Document and Process
For biomedical research, the Informed Consent Document Template prompts researchers to state whether they will return clinically relevant research results. There are template sections to choose from.
- If the results will not be provided to subjects, the template indicates that this is because the data, biospecimens or images will not be reviewed by a physician to determine the results.
- If the results will be provided to subjects, the template includes a statement that the subject may decide whether they want the information reported to them. There is an optional agreement section for the subject to initial whether or not they want the information.
The Informed Consent Document Template also includes template language and separate instructions for the return of genetic test results. These results can only be returned if they were performed in a CLIA-certified lab. The researcher must add an optional agreement section similar to the one for regular test results for subjects to indicate whether they want the genetic test results. The researcher must also state whether genetic counseling is available and who would assume the costs. All of these plans and procedures should be described in the Section VII.C.9 and VII.C.10 in the HawkIRB application.
References
- Wolf, S. M., B. N. Crock, B. Van Ness, F. Lawrenz, J. P. Kahn, L. M. Beskow, M. K. Cho, M. F. Christman R. C. Green, R. Hall, J. Illes, M. Keane, B. M. Knoppers, B. A. Koenig, I. S. Kohane, B. Leroy, K. J. Maschke, W. McGeveran, P. Ossorio, L. S. Parker, G. M. Petersen, H. S. Richardson, J. A. Scott, S. F. Terry, B. S. Wilfond, and W. A. Wolf. 2012. Managing incidental findings and research results in genomic research involving biobanks and archived datasets. Genetics in Medicine 14:61–384.
- National Academies of Sciences, Engineering, and Medicine. 2018. Returning individual research results to participants: Guidance for a new research paradigm. Washington, DC: The National Academies Press. doi: https://doi.org/10.17226/25094.
HawkIRB Automated Feature: Creating a ClinicalTrials.gov record (Push to PRS)
By Fozia Ghafoor, MBBS
To facilitate the ClinicalTrials.gov (CT.Gov) registration process, the HawkIRB application has a feature called “Push to PRS”. Research studies submitted under IRB-01 have the option of allowing the Protocol Registration and Results System (PRS) Administrator in the Human Subjects Office (HSO) to initiate the CT.Gov registration record on the behalf the Principal Investigator (PI). This article will describe this feature and how the UI researcher can request that this push of information be initiated.
HawkIRB includes a question that allows researchers to “push” IRB approved responses from the HawkIRB application to initiate a new CT.Gov record. This option applies to research projects where the protocol was developed and initiated by a UI Investigator and where the UI PI would be named as the responsible party in the PRS for a CT.Gov record. Clinical Trials eligible to push information from HawkIRB into a CT.gov record will be required to have a full developed protocol submitted by a UI PI.
How to initiate the push to PRS feature
When completing Section VII.B of the HawkIRB application, if the PI or their delegate indicate the UI Investigator is the one who initiated/provided the protocol in the response to question VII.B.2, an additional question will be generated (VII.B.12.c) that will ask if the PI would like the information provided in the HawkIRB application to be used to have a new CT.Gov record initiated on their behalf in the PRS.

If the PI agrees, a “Yes” selection to VII.B.12.c will pull information from the IRB approved application to initiate a CT.Gov record on the UI PI’s behalf. If the UI PI is not registered in the PRS system, the CT.gov Administrator will register the PI using their HAWKID and UI directory information. Upon registration, an email from the PRS system will be sent with login information to create an account in the PRS.
How the process works
The IRB will determine if the study meets the definition of an Applicable Clinical Trial (ACT), which requires registration and results reporting on CT.Gov. Studies which do not meet this definition may still choose to register for other reasons, such as publication requirements for some journals or NIH policy.
After the IRB has approved the project, the CT.Gov Administrator in the HSO will initiate the “push” of information from HawkIRB to the newly created CT.gov record.
The initial push by the PRS Administrator will auto populate only a portion of the required information. The PI or their delegate, called a Record Owner, will need to log into the PRS system to complete the rest of the CT.Gov record. Automated reminder emails will be sent from HawkIRB after 30 and 44 days for any incomplete CT.Gov records initiated on the PI’s behalf. All records must be completed in line with either FDAAA regulations, NIH policy, or ICJME requirements.
The table below represents a summary of which HawkIRB questions/responses auto populate fields in the new CT.gov record:
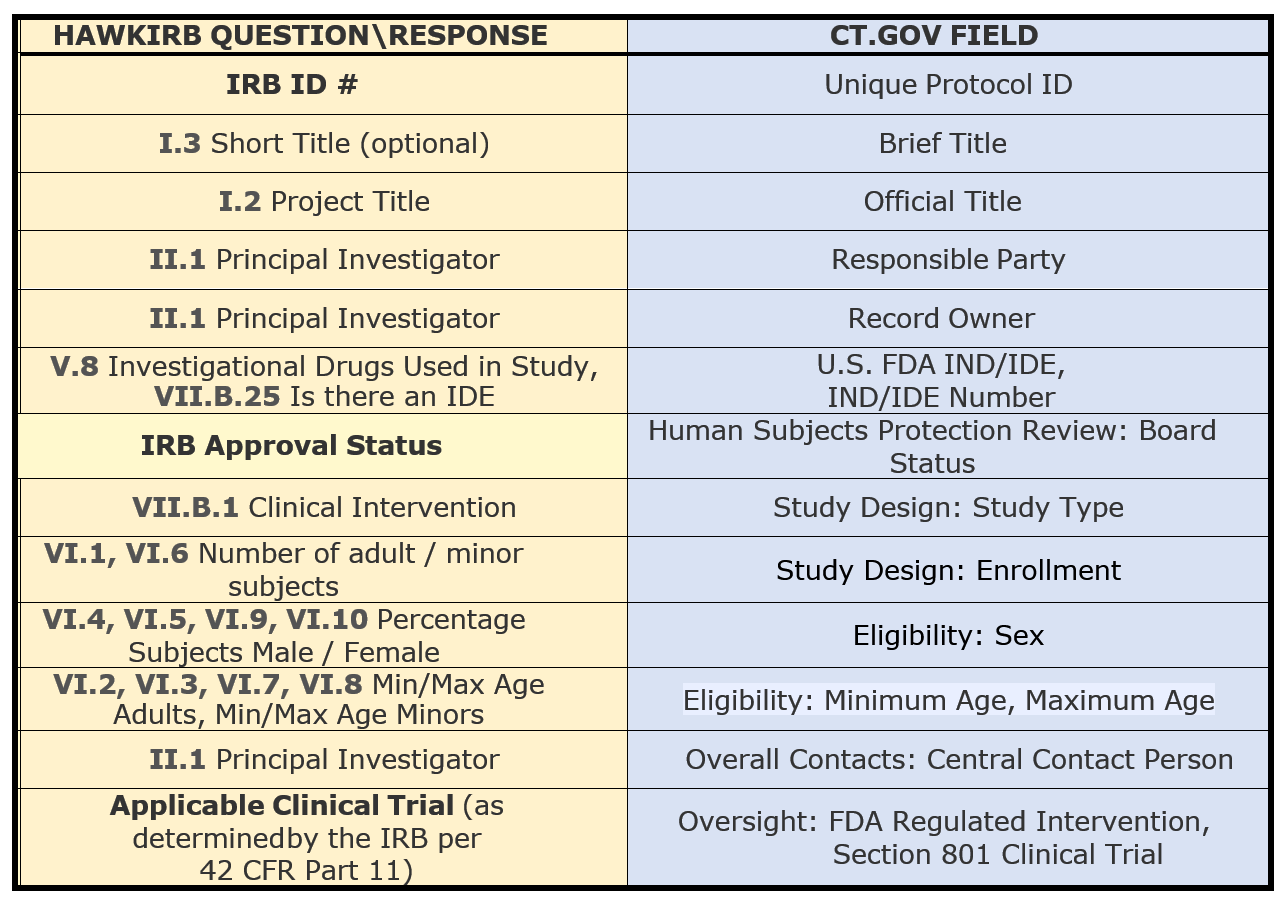
Project Summary page for a CT.Gov associated study
A HawkIRB application that is associated with a CT.Gov record will show CT.Gov related information on the Project Summary page. Under the FDA Regulated section, there will be an “Applicable Clinical Trial” listing with a Yes or No response as shown below.

For any CT.gov registered studies that are UI PI initiated, there will also be a new corresponding ClinicalTrials.gov tab on the HawkIRB Project Summary page. Selecting this tab will show information specific to the CT.Gov record associated with the IRB-approved application.
After selecting the ClinicalTrials.gov tab, the user will see basic PRS information as collected in the ClinicalTrials.gov record such as
- NCT Number
- Record Status
- Name of the Record Owner
- Primary Completion Date
- Study End Date
- Whether or not the study appears on a Problem Report
- Last update pushed from CT.gov to the HAWKIRB record
- A link to the full clinicaltrials.gov record
For additional assistance or questions about registering a clinical trial on ClinicalTrials.gov or using the PRS, please contact us at ct-gov@uiowa.edu or 319.335.6564.
IRB Advisor Newsletter, May and June 2021
By Kasey Lockett, BA
IRB Advisor (a publication of Relias) is a monthly newsletter with articles about issues facing IRBs, Human Research Protection Programs (HRPPs) and researchers. Current and past issues of IRB Advisor are posted in the “IRB ICON Course for Researchers” which is accessible to anyone with an active UI HawkID. The portal to this ICON Course is on the Education and Training page of the Human Subjects Office website.
This month we are spotlighting articles from the May and June 2021 IRB Advisor Newsletters.
Virtual Site Training Expands During the Pandemic (May 2021 IRB Advisor)
Although virtual training for clinical trial teams is not a new concept, it has gained traction during the Pandemic and the benefits are being widely embraced. Since anyone from a study site can attend a virtual training there can be a greater volume of attendance and greater training coverage. The on-demand nature of the training provides flexibility for sites to complete training at their own pace and at times that work best for them.
In addition to virtual training, virtual meetings between site staff and the study team are becoming more popular. These sessions allow site staff to voice their questions and hold a conversation with the study team. Plus, these meetings can be recorded and referred to throughout the life of the study.
Industry professionals refute concerns that connecting virtually is boring and claim that it can still be fun and interactive. Groups can include team building conversations and games within ‘live’ virtual trainings to keep participants engaged and learning. However, the choice between in-person versus online training should still be made strategically. For example, if the site conducting a clinical trial is new, it could be best to hold face-to-face meetings and build those new relationships.
There are major money saving benefits to virtual training as well. According to the senior director for training solutions at WCG Trifecta, statistics from a 9-month study show that delivering on-demand training reduces costs by 60%. This money can be invested into more training or other needs in the clinical development process.
Diversity in Clinical Trials Should Start with the Fundamentals (June 2021 IRB Advisor)
Diversity will only become more important as science and technology advance. In the future, algorithms utilizing genomic information will be used to make treatment decisions. Currently, 91% of genomic material available to scientists are of European ancestry. Designing clinical trials that match the population they are intended to treat is one of many steps to be taken toward more complete and equitable science. Diverse clinical trials begin with a commitment from leadership to address the topic within every aspect of the clinical trial process.
Conversations about diversity need not focus exclusively on race, as there are other groups that require a higher level of representation in clinical trials research. Underrepresented groups in clinical trials research include those that are not white, not wealthy, not male and those who do not live on the East or West coast in an urban or suburban center. In addition to those demographics, non-English speaking people, people with poor health literacy, older people, children, and adolescents are not properly represented. This means that researchers do not have proof of drug efficacy in these populations before the drug hits the market. This is an area in need of major improvement, as drugs have failed for a target population when they were tested on a different, non-representative population.
Professional scientists must consider a broad range of barriers to trial diversity. These barriers include, but are not limited to:
- lack of awareness of research opportunities
- deep mistrust of the healthcare system and research studies
- confusion and concern over what research is
- lack of plain language use in documents
- health insurance coverage
Barriers like these are viewed as downstream issues that need to be addressed at the inception of a research study. When a hypothesis is established, the corresponding protocol should be written with an inclusive angle.
One critical way to be inclusive is to think about where study subjects live, as ZIP codes often determine an individual’s health. Looking closely at inclusion and exclusion criteria is important as well. For instance, thinking about criteria that right be normal for people of color that could exclude them from a clinical trial for no real scientific reason.
Experts believe that good science is inclusive and to encourage that it would be best to handle it the same as other requirements, like well-made Data Safety and Monitoring Plans. When those plans aren’t present, there are punishments and scientists can lose the respect of their peers. Inclusivity and diversity in a protocol could become second nature if treated the same way. Bottom line, inclusiveness gives a market edge and results in better science for everyone.
Additional Articles in the May Issue:
- Pfizer and Moderna Begin COVID-19 Vaccine Trials in Younger Children
- IRB Staff Training Program Improves Consistency and Work Group Efficiency
- Reliance Teams Strengthen Relationships with Central IRB
- Exploitation Issues Arise in Study of Human Subject Incentive Payments
Additional Articles in the June Issue:
- The Role of Structural Racism in Lack of Clinical Trial Diversity
- The Issue of Using Race in Clinical Trials
- Clinical Trials with One Subject Raise Ethical Questions
- IRBs Experience Some Obstacles in Tweaking Reliance Programs
- Consistency Is a Chief Goal for Relying IRBs
In the News
- Why we remember more by reading – especially print – than from audio or video
- World's 1st multinode quantum network is a breakthrough for the quantum internet
- A new replication crisis: Research that is less likely to be true is cited more
- PARP inhibitor shrinks tumors in pancreatic cancer patients with mutations
- Landmark Alzheimer’s drug approval confounds research community

