Index
Two-Step Login Required for Research Administration Services
HawkIRB Enhancements & Initiating a Clinical Trials.Gov record on a UI PI’s behalf
FDA Updates Clinical Trial Guidance
OHRP Updates Decision Charts
What Is Not Considered Human Subjects Research?
External IRB Q & A: What Terminology Do I Need to Know?
Herky Hints: Subject Compensation
IRB Advisor, April and May
In the News
Two-Step Login Required for Research Administration Services
This message is a rebroadcast of an original message sent out by the Office of the Vice President for Research on July 15, 2020. Starting Saturday, August 1, 2020, Two-Step Login will be required to access the following electronic research administration services. This includes:
This change is part of an ongoing effort to enhance security on campus. Two-Step Login uses two-factor authentication to help verify identities of people logging in to UI systems. We recommend downloading the Duo Mobile app to complete your logins because it is safer, faster, and the most reliable way to complete two-step logins. If you’re currently using phone calls or other methods, switch to push notifications now.
For more information about Two-Step Login, visit the Two-Step Login support page.
Contact the Human Subjects Office (HSO) with questions
Email: irb@uiowa.edu or
Phone: (319) 335-6564
HawkIRB Enhancements & Initiating a Clinical Trials.Gov record on a UI PI’s behalf
Our ongoing efforts to streamline the human subjects research review processes have continued into 2020! The Human Subjects Office (HSO) is excited to inform you of the newest enhancement eliminating content duplication in the eResearch (HawkIRB) application and the ClinicalTrials.Gov registration process. Beginning in August 2020, research submitted under IRB-01 that is required or opts to register on ClinicalTrials.Gov (CT.Gov) is eligible for the HawkIRB system to generate a CT.Gov record on your behalf. There is a new question for researchers to “push” IRB approved responses from the HawkIRB application initiating a new CT.Gov record. This option applies to research with a developed protocol initiated by a UI Investigator. The UI PI would be named as the Responsible Party on a CT.Gov record for these instances. To launch this process, we made a few changes to how you answer questions in HawkIRB. See a detailed description of the HawkIRB changes on the Human Subjects Office website.
FDA Updates Clinical Trial Guidance
Joanie (Neyens) Hoefer, BS, CIP
On July 2, 2020 the U. S. Food and Drug Administration updated content to the question-and-answer appendix in its guidance titled “Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency” that was originally published in March 2020. The revisions to the guidance include the following summarized changes in the Appendix: Questions and Answers section:
- Clarification regarding two previously suggested methods for obtaining informed consent from hospital patients who are in isolation (Q10).
- A description of how to obtain informed consent where the potential subject can receive an electronic copy of the consent document but cannot sign it electronically or print it out to sign by hand. This only pertains to a study when the trial enrollment timeframe is limited, and the prospective trial participant must meet time-sensitive eligibility criteria (Q12).
- Clarification of previous recommendations for documenting the same information that would be documented during a face-to-face visit when using video conferencing for trial visits (Q20).
The entire guidance document is posted on the FDA website.
OHRP Updates Decision Charts
Joanie (Neyens) Hoefer BS, CIP
The Office for Human Research Protections (OHRP) recently published a new set of decision charts that are consistent with the requirements of the revised Common Rule that took effect in January 2019. These charts help investigators, the IRB and others determine if:
- an activity is human subjects research
- the research can be reviewed under expedited procedures
- the IRB can waive informed consent or documentation of consent
The new decision charts address the same issues as the previous version except they:
- do not include charts for determining if IRB review and IRB continuing review can be done on an expedited basis, and
- provide charts for the 2 new exempt categories.
OHRP cautions that the charts should only be used as a tool as they are generalized and may not be specific enough for certain situations. The text of the full regulations should be consulted when making final determinations regarding the review and approval of human subjects research.
Additionally, it is important to note that the decision charts only address the requirements found in 45 CFR part 46. They do not address the requirements of the subparts or other common rule regulatory agencies such as the Food and Drug Administration (FDA) and the National Institutes of Health (NIH).
What Is Not Considered Human Subjects Research?
By Maegan Tyrrell, BA
In a previous article, we went over the definitions of “research” and “human subject” as defined by the federal regulations. We also discussed how to ask if something needs IRB approval by using the Human Subjects Research Determination (HSRD) form. For this article, we will discuss what is not considered research as determined by the federal regulations (45 CFR 46.102). The first four sections below are specifically called out in the regulations as not being research.
It is important to note that UI faculty, staff, students and trainees could publish or present the results of activities that fall into any of these categories of ‘not human subjects research,’ even without IRB approval. The IRB only has oversight responsibility for activities that meet the regulatory definition of human subjects research (45 CFR 46.102). If you submit an HSRD form, you will have documentation that an IRB Chair determined that IRB approval was not necessary.
(1) Scholarly and journalistic activities (e.g., oral history, journalism, biography, literary criticism, legal research, and historical scholarship), including the collection and use of information, that focus directly on the specific individuals about whom the information is collected.
The main goal of these activities is to report factual, evidence-based information about the individuals involved and not to contribute to generalizable knowledge. These activities are not considered to be human subjects research because they focus on a specific individual and the activity’s findings are not extended to other groups or individuals. Case reports can fall under this category if the point of the report is to describe an unusual case and its outcome. According to UI policy, writing up the facts of three or fewer cases is not considered human subjects research and does not need IRB approval (UI IRB Standard Operating Procedures and Researcher Guide, Section I, Part 12.G).
(2) Public health surveillance activities, including the collection and testing of information or biospecimens, authorized by a public health authority. Such activities are limited to those necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance. Such activities include those associated with providing timely situational awareness and priority setting during the course of an event or crisis that threatens public health (including natural or man-made disasters).
Public health surveillance activities are not human subjects research because the Department of Health and Human Services recognizes that the requirements to conduct human subjects research (45 CFR 46) should not get in the way of a public health authority’s mission to protect and maintain the health and welfare of the public. An example of this would be the Iowa Department of Public Health setting up COVID-19 testing stations across Iowa.
IRB approval is required for public health research that is not authorized by a public health authority.
Conditions of public health importance according to the regulations include:
- Trends
- Signals
- Risk factors
- Patterns of diseases
- Increases in injuring from using consumer products
Examples of public health authorities:
- Local (Johnson County Public Health Department)
- State (Iowa Department of Public Health)
- Federal (Centers for Disease Control and Prevention)
3) Collection and analysis of information, biospecimens, or records by or for a criminal justice agency for activities authorized by law or court order solely for criminal justice or criminal investigative purposes.
Criminal justice investigations typically do not meet the regulatory definition of human subjects research.
(4) Authorized operational activities (as determined by each agency) in support of intelligence, homeland security, defense, or other national security missions.
National security activities conducted by, or on behalf of, a federal agency also do not meet the regulatory definition of human subjects research.
Additional Activities That Typically Do Not Need IRB Approval
The next three activities do not meet the regulatory definition of human subjects research, but they are not specifically mentioned in the federal regulations. With all three of these activities there could be interviews and surveys involved (standard research methodology), but if the information gathered is used for non-research purposes, it does not need IRB approval.
Implementation of a proven intervention in a clinic, school, other setting. A proven intervention is something that previous research or experience supports as an effective way to do a particular activity. This is referred to as implementation of evidence-based practice. For an intervention to fall under this category, it cannot be done for research purposes and any findings from the intervention would not contribute to generalizable knowledge. For example, if a clinic wanted to implement a proven intervention to lower infection rates as part of standard of care operations, it would not be research because they implemented a strategy that has evidence behind it to show it works. In this case, the purpose of implementing this intervention is not to further scientific knowledge in a particular field of study. The purpose or intent of the project is to reduce the infection rate for patients in the clinic.
Needs assessment in a specific population. The purpose of a needs assessment is to identify and meet the needs of individuals or a particular population, not to further scientific knowledge. The federal regulations state that a project is defined as being research when it is designed to develop or contribute to generalizable knowledge. A needs assessment generally does not fit this criterion.
Quality Assurance/Quality Improvement (QA/QI) Projects. The main purpose of these projects is to evaluate or improve the delivery of a pre-existing service or program. For example, the patient satisfaction surveys that UI Health Care sends out after a visit is intended to help departments improve the patient experience. They are not used to answer a research question or hypothesis or to further scientific knowledge in a particular field of study.
Summary
This article provides broad examples of activities that are not considered research, but this is not an exhaustive list. Every project is different. If there is any doubt about whether a project needs IRB approval, the researcher should submit an HSRD form to ask for a determination about whether the project meets the regulatory definition of human subjects research. For more elaboration on this topic, please visit the Human Subjects Office’s page about the HSRD form.
External IRB Q & A: What Terminology Do I Need to Know?
By Jarrod Feld, BA
This is the first in an ongoing series of monthly articles about using an IRB of record that is outside the University of Iowa.
The final component of the federal regulations for the protection of human subjects that took effect in January 2020 (45 CFR 46.114) requires the use of a single IRB of record for most multi-site studies with federal funding. Additionally, many corporate sponsor-funded studies choose to utilize a single IRB of record for multi-site drug and device studies. For reference, exemptions from the single IRB model exist and were described in the January 2020 IRB Connection Newsletter.
We frequently field questions from researchers and study coordinators about when a single IRB is needed, and even more frequently, about the terminology used in the single IRB realm. In this series of articles we will describe the terms used in multi-site studies, explain the differences between the various IRB options, discuss reliance and workflow expectations for the various study sites, explore various multi-site models, and hopefully help researchers better understand how these differences affect what is required locally, from other sites, and from other IRBs.
First let’s look some commonly used terms and their meanings:
Terminology
- Central or Single IRB: These terms are used interchangeably. Both terms indicate that a single IRB will be oversee a multi-site study. We typically use the term “single IRB model” since that language is used in the federal regulations. A single IRB is the IRB of record for any multi-site study that is required (or chooses) to utilize this model. The term “central IRB” typically indicates the IRB of record for a network or consortium working together on multiple studies.
- External IRB: This is any non-UI IRB that oversees UI research activities for a multi-site study. An external IRB might be at another academic institution, or it may be a commercial, for-profit IRB, such as Western IRB (WIRB) or Advarra. These are commonly referred to as “commercial IRBs.” Generally, when an academic institution IRB serves as the IRB of record, researchers at that institution are conducting the same research under the same protocol that is being conducted at the University of Iowa and other sites. Commercial IRBs do not have their own researchers or a Federal Wide Assurance (FWA); they only serve as the IRB of record.
- Lead vs Relying: In a multi-site study using the single IRB model, the sites and/or sponsor (or granting agency) choose one IRB to serve as the IRB of record, or the lead IRB. When they choose the UI IRB, we are the lead IRB, while other study sites are relying sites. Conversely, when another entity serves as the single IRB, Iowa is a relying site. However, a lead site does not necessarily have to be the IRB of record (or lead IRB). For example, one institution may provide the protocol for all study sites but choose another institution or entity to serve as the IRB of record. More detailed information about lead and relying sites and IRBs will follow in subsequent articles in this series. Note that these terms are not typically used when there is a commercial IRB of record, as all sites are relying on that IRB.
Because each study is different, and since the various commercial and external IRBs have their own requirements and processes, we recommend reaching out to staff at the Human Subjects Office for guidance as soon as you know that the UI will participate in a study using a single IRB model. Additional information and contact information is located on the Central and External IRB page of the Human Subjects Office website. Email contact is uirb-external@uiowa.edu.

Herky Hints: Subject Compensation
By Joanie (Neyens) Hoefer, BS, CIP
Providing compensation to subjects is a common and generally accepted practice. There are many factors to consider when deciding the amount of compensation. It is very important to document your compensation plan clearly in the HawkIRB application.
IRB Policy
University of Iowa IRB policy states that compensation should be based on the amount of time a subject has spent participating in the study, loss of wages or other inconveniences incurred. It can also include reimbursement for expenses that the subject incurred during their participation. (UI IRB Standard Operating Procedures (SOP) & Researcher Guide Section II, Part 12.G) Compensation must not be withheld until a subject completes the entire study but should be provided on a pro-rated basis—either per study visit or per study procedure.
UI Policy
All UI faculty, staff, and student researchers must comply with the UI cash handling and research subject compensation policies when providing compensation to subjects. If you are providing cash or cash-equivalents to subjects, an approved cash handling policy must be on file with UI Accounting Services before the IRB will approve your study.
You must collect some basic information about subjects when providing compensation. This includes their name, mailing address and, in some cases, Social Security Number (SSN). This information allows the UI to meet government reporting obligations. If the research subject is a non-resident alien, the UI is required to withhold 30% from the payment.
You only need to collect the subject’s SSN for payments that are more than $100 for a single payment OR if the subject is expected to receive cumulative payments of greater than $600 for the calendar year. The UI policy regarding collection and retention of SSNs is in the UI Operations Manual, Chapter 36.
HawkIRB Application
You will describe your compensation plan in Sections VII.E.9-19. First, indicate if subjects will be compensated (Section VII.E.9).
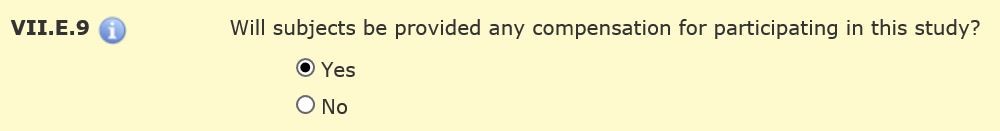
Then specify the type(s) of compensation:
| Cash (Section VII.E.10) |  |
| Gift cards (Section VII.E.11) |  |
| Check (Section VII.E.12)—Indicate whether the researcher or Accounting Services will provide the check to the subjects in Section VII.E.13. |  |
| Other (Section VII.E.16)—Mark “Yes” if you are providing any other compensation, such as meals, parking, course credit/extra credit or non-monetary items of value such as notepads, pens, t-shirts, small toys, or other gift items, etc. Describe this compensation in Section VII.E.17. | 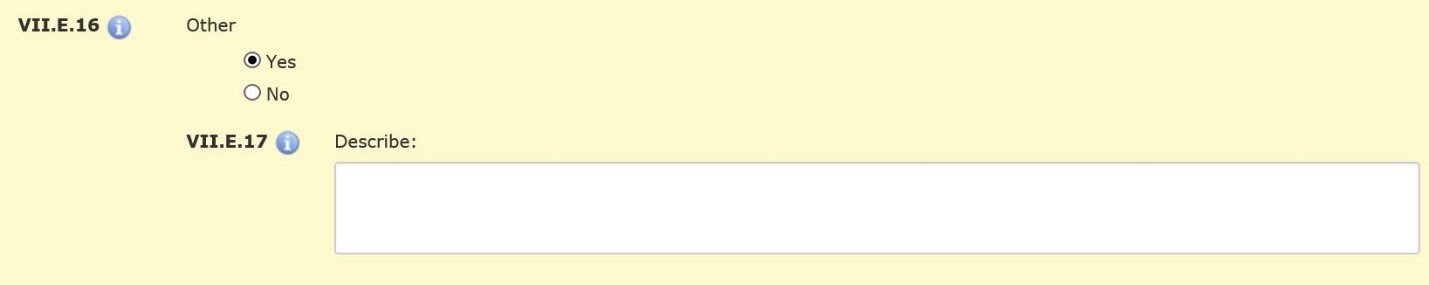 |
Section VII.E.18 will open if you are providing compensation that requires a cash handling plan to be on file with Accounting Services.
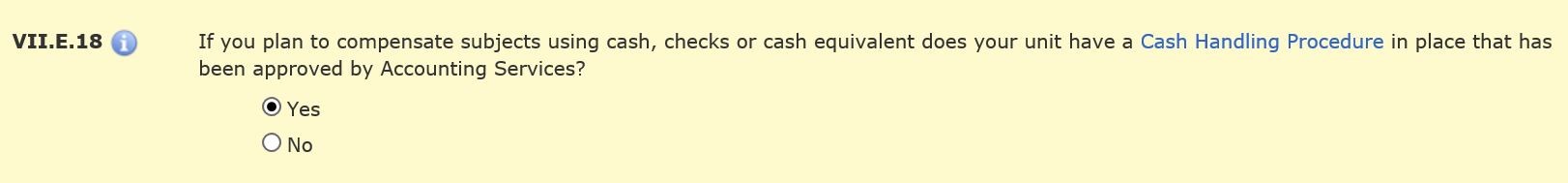
You do not need to attach the actual cash handling plan to the HawkIRB application. Just attach documentation of approval of the plan. The attachment could be a screen shot of the routing history for the cash handling plan through Universal Workflow.
In Section VII.E.19, provide a thorough, detailed description of your research subject compensation plan.

Include additional information for the following compensation methods:
- Gift cards - Specify the dollar amount of the card and the gift card merchant.
- Subjects compensated by a study sponsor or a non-UI entity (i.e. mTurk, lead site of a multi-site study, etc.) - State who will provide the compensation and the method for how it may be provided.
- Extra credit to student subjects –
- Provide an alternative extra credit option for students who do not want to participate in the study. More information about providing extra credit to student participants can be found in the UI IRB Standard Operating Procedures (SOP) & Researcher Guide (Section II, Part 7.K.i).
- Provide a complete description of the process for reporting the extra credit to the course instructor. The instructor should only be informed of the amount of credit that a student is receiving.
If you are required to collect SSNs for compensation purposes, indicate this in Section X.2.
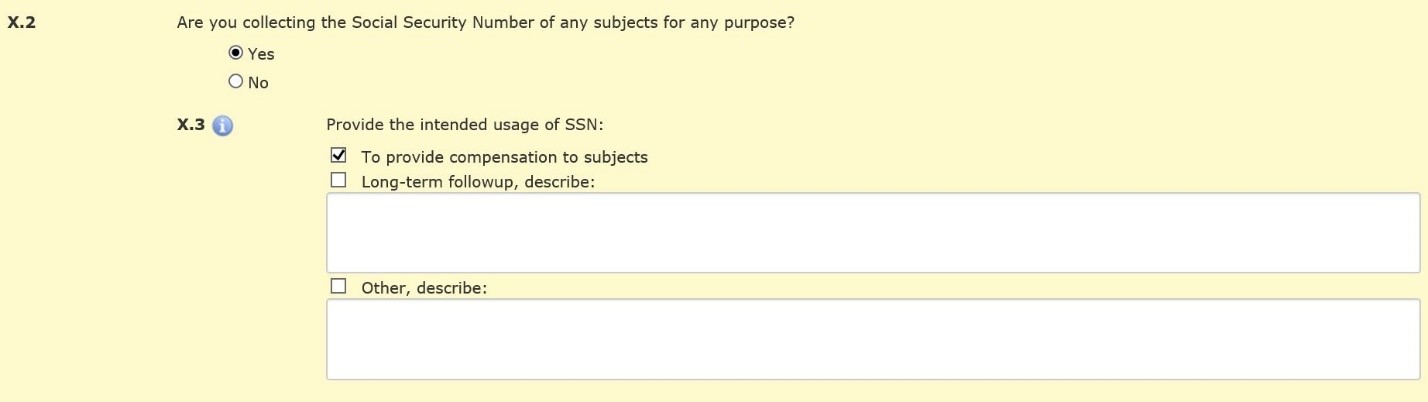
Then describe your plan to keep this information confidential in Section X.4, even if you do not intend to retain the SSN number.

IRB Advisor, April and May
By Maegan Tyrrell, BA, CHES
IRB Advisor (a publication of Relias) is a monthly newsletter with articles about issues facing IRBs and individuals involved with human subjects research. Current and past issues of IRB Advisor are posted in the “IRB ICON Course for Researchers” which is accessible to anyone with an active UI HawkID. The portal to this ICON Course is on the Education and Training page of the Human Subjects Office website.
This month we are spotlighting articles from the April (Interactive Online Checklists Help Investigators With Informed Consent) and May (What Happens to Human Research in the New Pandemic Era?) 2020 IRB Advisor Newsletters.
Interactive Online Checklists Help Investigators With Informed Consent
The University of Utah replaced almost all their templates with an informed consent checklist. The checklist is similar to internal checklists used by the University of Utah IRB to prescreen applications. The IRB made this change to better explain the elements of consent and key information that are required by the federal regulations under the revised Common Rule. Researchers at the University of Utah praised the checklist formatting after it was rolled out. One of the biggest benefits of providing this checklist is that investigators have improved their submissions, resulting in fewer revisions. The decline in revisions to applications saves IRB staff time and allows for more efficient reviewing processes.
What Happens to Human Research in the New Pandemic Era?
A question on many minds currently is how will human subjects research look after COVID-19? Will there be a new normal? Jill Johnston, president of study planning and site optimization at WIRB-Copernicus Group (WCG) noted, “We are not going to return to normal. It will be a new and improved normal. There is more acceptance of virtual trials and telemedicine.” While the switch to going virtual was necessary, it has proved to be an effective way to get things done from study visits to IRB meetings. The University of Cincinnati IRB always had IRB meetings in person, but after being forced to start holding virtual meetings they realized how well this option works. An IRB chair from the University of Cincinnati indicated plans to use this process more moving forward. Other IRBs, like Advarra, already held all meetings virtually, so the switch was less of an issue for them. A member of Advarra’s team stressed the importance of IRBs having a plan in place in the event that normal operations are not possible. Researchers are also integrating virtual activities into study visits and clinical trials. James Riddle from Advarra stated that virtual trials increase access for people who could not attend in-person clinic visits before. This expands the number of people who could potentially participate in a clinical trial.
Read the rest of these articles to learn more about virtual considerations for IRBs and investigators and how informed consent checklists are changing the way researchers approach writing informed consent forms.
Other articles in the April issue include:
- COVID-19’s Effects Hit Healthcare, Research Institutions
- Real-Time IRB Process Reduces Turnaround by 71%
- IRB’s Re-Engineered Program Makes It More Responsive
- Study Reveals Preferences for Simpler Research Language
- New Working Group to Produce Guidance for Pediatric Gene Therapy
- IRBs Can Prepare for Cannabis Research
Other articles in the May issue include:
- FDA Guidance Offers Foundation for IRB Researcher Flexibility
- Second Phase of Pandemic Raises More Questions, Concerns for IRBs
- Shortcuts in Clinical Trials May Cause More Harm Than Good
- Unique Ethical Concerns for Study Participants in Neuroscience Research
- Researchers Identify Ethical Concerns with Pragmatic Trials
In the News
- Six months of coronavirus: the mysteries scientists are still racing to solve Nature
- A Shot to Protect Against HIV The New York Times
- The gut microbiome of Irish Travellers gives a timely public health lesson Medical Xpress
- COVID stress syndrome: Concept, structure, and correlates Wiley Online Library

