IRB Education for your Group: Presentations to Suit your Needs
By Brent Collinsworth
The University of Iowa Human Subjects Office provides many educational resources to the UI research community. As a reminder for the Spring 2017 semester, we want to highlight a resource that may be especially useful to instructors of research methods courses, other research-related courses, journal clubs and orientation sessions or seminars for new faculty, graduate and professional students. The IRB Education and Outreach program is available upon request to give classroom and group presentations about the ethical conduct of research and the UI IRB review process. We can tailor the content to suit your needs, and we are able to present to a wide variety of courses and groups, including evening and online classes.
We offer a general IRB overview presentation that provides a broad overview of topics that are relevant for students and others who plan to conduct research themselves, whether as a course requirement or as an honors, Master’s or doctoral dissertation. This presentation can cover some or all of the following:
- The definition of human subjects research
- How to ask if you need IRB approval
- Ethical conduct of research (the Belmont Report)
- The UI IRB review process
- Informed consent
- Research off campus or outside the United States
- Course-related student projects
- IRB Resources that are available for UI/VA researchers
This presentation provides a good orientation to the UI IRB and the IRB approval requirements for faculty, staff and student researchers. We also offer a full presentation about the ethical conduct of research. It includes an in-depth discussion of the ethical principles of human subjects research that are outlined in the Belmont Report and examples of unethical research conducted both historically and recently. This presentation is designed to help students apply basic ethical standards to real-world examples of human subjects research. It would be an excellent supplemental lecture for any research methods course.
Each of these presentations typically fills a 50-minute class period, but we can tailor the length and topic to suit the needs of the class or group. Contact us at irb-outreach@uiowa.edu to request a presentation.
Documenting subject agreement for optional research activities.
By Laura Dallas
If your study obtains written consent from subjects, one signature at the end of the consent document is normally sufficient to document the subject’s agreement to participate, even if the research contains more than one procedure or test. But in a treatment study with optional study procedures that use protected health information (PHI), the consent document must separately document the subject’s agreement to participate in the study and their choice about the optional (non-treatment) procedures.
For any study that has optional procedures, researchers should ask subjects to initial a “yes” or “no” choice for each procedure in the Informed Consent Document. It puts “on the record” the subject’s decision, which is especially helpful when the optional procedure involves the researcher taking additional measurements or doing additional activities after the study visit. Some examples include: sharing the subject’s data with other researchers or running additional tests on a biological sample. For optional research components like these, having the subjects wishes documented is helpful because one can go back to the consent document and check what was recorded if there is ever a question regarding whether permission was granted by the subject.
There are some instances where it may not be necessary to document the subject’s choice to participate in an optional research procedure. For example, if the subject can simply decide to take part in the optional procedure or not. In particular, these might be situations where it is not necessary for a decision to be made at the time of consent, and it is the subject who takes the initiative to participate. An example of this is attending an optional focus group at the end of the study, or completing an additional optional survey about the research experience.
But when the study involves a treatment and the researchers use or create protected health information (PHI), documenting the subjects choice for optional study activities is mandatory. For this particular type of study there is a regulatory requirement (CFR 45 part 164.508(b)(3)(i)) to obtain documentation from subjects that they have “opted in” to an optional research procedure.
The HIPAA privacy regulations require that subjects give researchers permission to access or create PHI. This requirement is met by language in the consent document section “Will my health information be used in this study?” Signing the Informed Consent Document indicates the subject’s authorization to the creation and use of protected health information for the purposes of the research.. The regulations allow this authorization to be combined with the research consent document, and have one signature serve as the subject’s agreement for both participation in research, and the release of PHI for use in the research.
Requirements change further if the research study provides treatment to the subject and the study has optional components that are not directly related to the research treatment. An example of this would be an investigational cancer treatment study that also proposes collecting blood and health data for future research on cancer. In that case you must always provide the subject the chance to document an “opt in” to the optional components by initially or checking a “yes” choice for the procedure. Under the regulations, you cannot ask for a subject’s authorization to release PHI related for their treatment (even research treatment) and at the same time require the release of PHI for other purposes not related to the treatment. This may compel someone to agree to release their PHI for optional research, because they are motivated to participate in the research treatment. In this situation, the subject must be given a clear choice to opt in to the optional research components that require the use of PHI.
Luckily, researchers can satisfy this requirement to document the subjects’ agreement with a simple paragraph explaining what is optional and an area to clearly designate the subject’s choice to opt in. The IRB will require this to be added if it is not included in the Informed Consent Document when the HawkIRB application is submitted.
Herky Hints: Mods Don’t Stop the Research
By Sarah Heady
Everything you think you know about submitting modifications is wrong.
Okay, I’ll confess to exaggerating to get your attention. But some UI faculty, staff, and student researchers mistakenly think that submitting a modification to the Institutional Review Board (IRB) means all research comes to a screeching halt until the modification is approved. WRONG. SO NOT TRUE. NOPENOPENOPTOPUS. When a modification is submitted, the PI must wait for IRB approval to implement the changes described in the mod (you don’t need to see modification spelled out every single time I type it, right?). But all previously approved aspects of the research project keep on trucking. Now of course this isn’t always black and white. Consider subject enrollment—if there are new risks or benefits for potential subjects to consider, or new study procedures you want to implement with currently enrolled subjects, you should not enroll new subjects until the modification is approved.
Changing Research Procedures (Adding, Removing, and Editing)
When out and about and monitoring research projects, I frequently find HawkIRB applications with outdated information about project procedures, protocols, or people (deactivated research team members are very common). It’s very important to keep the HawkIRB application up to date. When a HawkIRB New Project form is submitted to the IRB, the PI agrees to never, ever change any aspect of the research without prior approval (minus one critical exception—in the limited cases when subject safety is affected, researchers can change study procedures and then submit a HawkIRB mod form). The PI makes the following assurance to the IRB:
-
I will not implement any changes in the approved IRB application, study protocol, or informed consent process without prior IRB approval (except in an emergency, if necessary to safeguard the well-being of a human participant).
When you need to change anything about the study design, procedures or any of the IRB approved study documents, submit the modification, get IRB approval, and THEN implement the changes.
Frequent Modifications
Changing the research team
For a simple research team edit—simple meaning: adding or removing a non-PI research team member—the HSO usually reviews and approves these modifications within 24-48 hours. The new team member cannot work directly with subjects or access subjects’ identifiable private information until the mod form is approved. But everyone else on the research team can keep researching and working on the study.
Updating funding sources
When adding or removing a funding source, consider how the funding will affect research procedures. If procedures do not change, then research can continue while the mod is going through IRB review. Any procedures that are added due to the new funding source should only be implemented once the modification is approved. And of course,, you should describe the new procedures in the mod form in addition to adding the new funding source.
Adding a subject population
When a new subject population is added to a research project, work with the previously approved subject group(s) can continue, but you can not implement any research activity with the new population until the mod form is approved. The research procedures haven’t changed (in this example) but you have to wait for approval to work with the new people (the new subject population). Again, if there are new recruitment methods or other study procedures with the new subject population(s), they should be described in the mod form.
Need a quick look at your changes
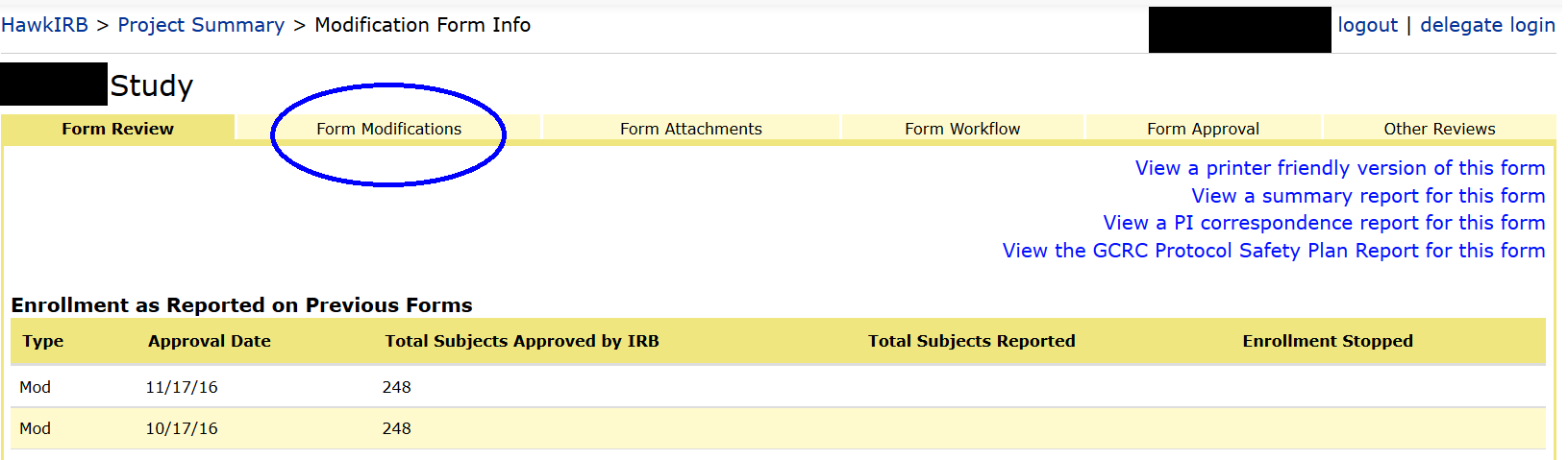
Check out the split screen history HawkIRB offers. There are multiple ways to access this view. You can look back to see what was changed in a previous mod form. Open the Project Summary page and click on a mod form in the History section in the lower portion of the screen. Under ‘form review’, select the ‘Form modifications’ tab. HawkIRB will display a split screen image, with the changes you made (New Value) in the right hand column, and the previous content (Old Value) on the left.
Prior to submitting a mod, HawkIRB will ask you to review the edits; the message reads “Review the modifications that have been made on this form before submission.” This is the last stop before you click the ‘submit form’ button. Make sure everything you wanted to change shows up here.
More information is available at our HawkIRB training sessions, in the Modification FAQ. Or as always, contact us directly by phone (319-335-6564) or by e-mail (irb@uiowa.edu). Any questions?
The IRB Advisor Newsletter, February 2016:
The Ethical Question of Denying Children Antibiotics
Is it safe and ethical to conduct research that alters the standard schedule for antibiotic use in very young children? Due to the overuse and misuse of antibiotics, antibiotic resistant bacterial infections are more common, and diseases that once were easily treatable are now becoming harder to manage. This raises questions about whether it is possible to give fewer antibiotics for a shorter duration without affecting treatment quality. The National Institute of Allergy and Infectious Diseases at the National Institutes of Health (NIH) is sponsoring a clinical trial of 400 children, age 6 months to six years, with community-acquired pneumonia who will be randomly assigned to receive either the standard 10-day regimen of antibiotics or a shortened 5-day regimen. This article includes an interview with one of the principal investigators of the study, Dr. C. Buddy Creech. He discusses the ethical issues with this type of study, and how researchers addressed them in the study design prior to IRB submission at the five study sites.
Read the full article in the February issue of IRB Advisor.
The December, January and February issues of IRB Advisor include several articles that may be of interest to UI researchers. Here are just a few:
The 21st Century Cures Act Easily Passed, But is it Good for Research Protection? (February)
People with Mental Illness Often Excluded from Clinical Trials (January)
Clinical Trial Addresses the Tricky Process of Revealing Genetic Risk Factors for Alzheimer’s (January)
NIH Designates LGBT Community as Health Disparity Group (December)
When IRBs Take a Walk on the Wild Side: The Dark Web (December)
What is IRB Advisor?
IRB Advisor is a monthly newsletter that contains articles about regulatory issues, informed consent, current events in human subjects protections as well as articles about IRB administrative and management issues. The UI IRB subscribes to this publication as a resource for UI faculty, staff and student researchers as well as for IRB members and Human Subjects Office staff. Each month the IRB Connection Newsletter features an article from the current issue of IRB Advisor.
Current and Past Issues
There is a link to current and past issues of IRB Advisor on the Education and Training page of the Human Subjects Office web site. This link provides automatic access to the newsletter from all computers with a University of Iowa IP address.
The University of Iowa username and password cannot be posted on the Human Subjects Office web site. UI faculty, staff and students and VA researchers may contact the Human Subjects Office to request the username and password to access IRB Advisor from a personal computer. Contact us by e-mail (irb-outreach@uiowa.edu) or call us at 319-335-6564.
Continuing Education Credits
Individual newsletter subscribers can receive 1.5 AMA PRA Category 1 Credits™ or 1.5 nursing contact hours for reading an issue and completing an online test. However, since this is an institution-level subscription, UI researchers must purchase access to an individual issue for $40, or purchase a full subscription, to receive CME/CE credits. Visit the AHC Media web site for information about subscription options.

