About ClinicalTrials.gov
ClinicalTrials.gov is a public database containing information about federally and privately supported clinical trials for an array of diseases and conditions. A service of the U.S. National Library of Medicine (NLM) at the National Institutes of Health (NIH), and in collaboration with the Food and Drug Administration (FDA), ClinicalTrials.gov requires registration and results reporting for Applicable Clinical Trials (ACT) and clinical trials wishing to share their findings with the public.. Background information regarding history, policies, and laws can be found on ClinicalTrials.gov Section 801 of the Food and Drug Administration Act (FDAAA 801) specifies registration and results reporting requirements for clinical trials (for full documentation describing the law, see FDAAA 801). Investigators wishing to publish on ClinicalTrials.gov must do so in the Protocol Registration and Results System (PRS) at register.clinicaltrials.gov. The purpose of this page is to help investigators at the University of Iowa (UI) understand their responsibilities for ClinicalTrials.gov registration and results reporting.
ClinicalTrials.gov Investigator’s Guide
New as of 02/06/2024: The ClinicalTrials.gov Investigator's Guide has been updated. The ClinicalTrials.gov Investigator’s Guide covers the University of Iowa’s policy related to ClinicalTrials.gov registration and results reporting. This documentation satisfies the NIH requirement for data sharing, as well. A clean and tracked changes version are included to see recent changes. A summary of the recent updates is below:
- Section 8.0.3 Potential Consequences of noncompliance has been updated; The Civil monetary penalties can be charged up to $14,262 per day.
Final Rule Update
The U.S. Department of Health and Human Services has issued a final rule (42 CFR 11) about ClinicalTrials.gov. For more information, please see the HHS news release and NIH Office of Science Policy blog post. The Final Rule became effective on January 18, 2017, and required all institutions to comply no later than April 18, 2017. Institutions found in violation of this rule could be subject to civil penalties of up to $14,262, as well as an additional $14,262 per day, if the issue is not resolved within 30 days. Additionally, funding to the institution can be reduced or stopped and non-compliance could impact future funding for the institution. Besides civil penalties, criminal penalties are also possible for investigators found in non-compliance of this rule. A checklist-based tool to assist responsible parties in evaluating whether a study is an Applicable Clinical Trial based on 42 CFR 11.22(b) is now available here.
Help and Resources
For investigators who are new to the registration process at ClinicalTrials.gov, or who are struggling to understand how to use the system, there are several resources available(see ‘Resources’ link below). The ClinicalTrials.gov Checklist can be used to help any investigator get started and keep track of ongoing expectations related to the ClinicalTrials.gov Protocol Registration and Results System (PRS). Slides from an ICTS presentation are available for registering a record and expectations for record maintenance. The PRS administrator, who oversees all records for the University of Iowa, provides ongoing education sessions throughout the year. Both the PRS administrator and the IRB also assist investigators by providing communications regularly regarding the application of federal regulations, providing notification when a record update is required, and by informing the Responsible Party when their record is in non-compliance with federal regulations. Additionally, several departments have departmental liaisons who serve on a ClinicalTrials.gov working group. These liaisons are fully trained in registering a record and submitting results and can assist their departments in completing their ClinicalTrials.gov PRS records. More information on the ClinicalTrials.gov working group can be found here.
Need Help?
Fill out the form here to request help from a ClinicalTrials.gov Departmental Liaison
a. Who is required to register?
b. When must I register on ClinicalTrials.gov?
c. What should I do after registration?
d. Getting Started - ClinicalTrials.gov Checklist
2. Applicable Clinical Trials (ACT)
4. NIH and ICMJE Reporting Requirements
5. Results Submission and Required Documents
6. Observational and Expanded Access Studies
7. Protocol Registration and Results System (PRS)
b. Veteran's Affairs (VA) Studies
11. ClinicalTrials.gov Office Hours
13. ClinicalTrials.gov Working Group Departmental Liaisons Page
1. Registration
Registration is required for all Applicable Clinical Trials (ACT) based on the federal regulations (42 CFR 11). However, other agencies have adopted their own policies in regards to ClinicalTrials.gov requirements. Studies utilizing NIH funding or wishing to publish in an International Committee of Medical Journal Editors (ICMJE) may also be required to register on ClinicalTrials.gov, even if the study does not meet the criteria of an ACT. Criteria for determining if a study meets registration requirements can be found on the website of each organization. Additionally, the University of Iowa IRB assists investigators in making determinations regarding registration requirements during their review.. To register a study, investigators must use the Protocol Registration and Results System (PRS) at www.register.clinicaltrials.gov. To get started, investigators should contact a PRS administrator by emailing ct-gov@uiowa.edu to have an account created for them. Once registered in the system, users will be able to add new projects in the PRS at any time. The University of Iowa stores and maintains their own records for ClinicalTrials.gov, but the review and publication of these records to ClinicalTrials.gov happens outside the institution by the PRS team.
a. Who is Required to Register?
Clinical trials registration and results reporting is required by law for all Applicable Clinical Trials, for clinical trials funded by NIH, and for investigators wishing to publish trial information in an ICMJE journal. At the University of Iowa (UI), the Responsible Party of a clinical trial is the person who sponsors, or initiates, the trial. If this is a UI investigator, the term ‘Sponsor-investigator’ identifies this role in the record. For trials sponsored by an industry sponsor, the industry sponsor is required to register and maintain the study record and must provide the NCT number to the UI Principal Investigator (PI). In the PRS, the term ‘Principal Investigator’ defines a situation where the sponsor of the study designates a University of Iowa Principal Investigator as the responsible party or the record. If the sponsor is not a University of Iowa faculty or staff member, but is an investigator at another institution, the responsibility of reporting to ClinicalTrials.gov falls upon the sponsor’s institution to register and maintain the record.
If one of the following examples applies to you or your study, you are considered a sponsor-investigator:
- You hold the Investigational New Drug Application (IND) or the Investigational Device Exemption (IDE) for your study (responsible party = sponsor-investigator);
- Your study involves the UI serving as the coordinating center for a clinical trial under a prime award (as opposed to a subcontract) and the trial is investigator-initiated;
- You do not hold the IND or IDE for your study, but another UI investigator does and is not on your research team;
- The clinical trial is regulated by the FDA (phase II through IV trial) and involves a drug, biologic or device, but does not require an IND/IDE; and/or
- Your study involves a clinical trial and has NIH funding.
For studies that have the UI as the IRB of record, the HawkIRB application captures trial information and NCT# in Section VII.B.1. Entering a NCT# is required for Phase 2-4 studies and is currently optional for Phase 1 studies and early device feasibility studies unless NIH funding is used.
Find more information at ClinicalTrials.gov under "Who is Responsible for Registering Trials and Submitting Results?"
b. When must I register on ClinicalTrials.gov?
Applicable Clinical Trials (ACTs) and NIH funded trials are required to register on ClinicalTrials.gov within 21 days of enrollment of the first subject. Throughout the life of the record, updates must be made anytime the study plan changes, typically within 30 days. Updates are required at least every 12 months, even if nothing has changed. Additional information can be found on ClinicalTrials.gov (“Which Trials Must Be Registered and Have Results Submitted to ClinicalTrials.gov?”). Trials planning to publish in in ICMJE journals must register a study on ClinicalTrials.gov prior to enrolling the first subject.
Below is a table indicating when registration requirements apply:
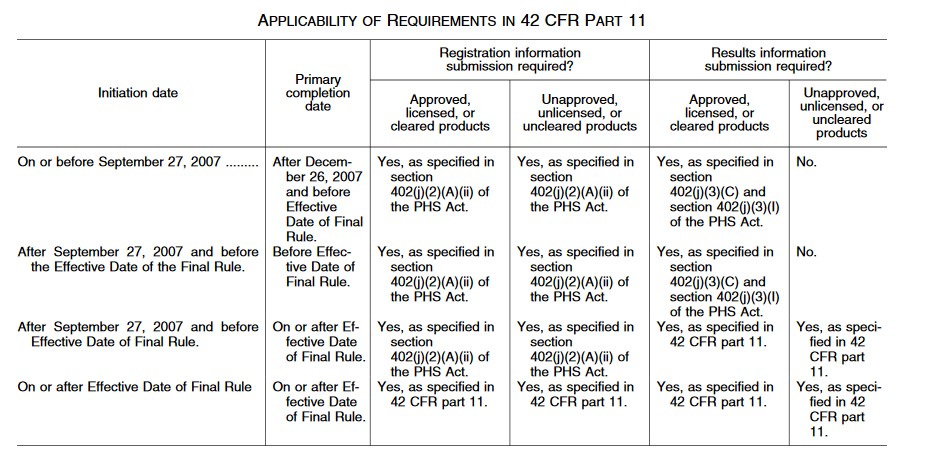
c. What Should I Do After Registration?
Registering in the PRS on ClinicalTrials.gov is only the first step. FDAAA 801 and 42 CFR 11 require reporting of results, adverse events (expected and unexpected), as well as modifications or updates to the study. The record must be updated anytime the study procedures change, or when enrollment ends or is suspended. Additionally, at the time results are ready to be posted, the study protocol and Statistical Analysis Plan (SAP) must also be included with the record. The University of Iowa IRB will facilitate the process of informing sponsor-investigators when updates are required by during their review of the IRB application. Even if nothing has changed in the study, the record requires yearly review and approval, even if nothing has changed. The Responsible Party or Record Owner (individual responsible for the record’s content) may update the record at any time, but investigators are recommended to make any needed changes after submitting IRB review. The IRB will direct the investigator on any needed changes, and may require changes to the protocol in their own review.
d. Getting Started - ClinicalTrials.gov Checklist
A tool is available for University of Iowa investigators planning to register or report results on ClinicalTrials.gov. The tool, ClinicalTrials.gov Checklist helps investigators determine when a specific regulation may apply to their study, what materials are needed to register and report results, and provides links to a number of useful planning materials.
As of 09/01/2022 ClinicalTrials.gov checklist has been updated. Here is a summary of the new version's content.
- Applicable Clinical Trial criteria
- Registration requirement on the basis of NIH funding
- ICMJE requirements
- ClinicalTrials.gov registration requirements
- Different Scenarios for ClinicalTrials.gov registration
- Basic steps for ClinicalTrials.gov registration
- Requirements for results submission with checklists, templates, and examples
- Required documents for results submission
- ClinicalTrials.gov good cause extension request deadline and checklist for required information
2. Applicable Clinical Trials (ACT)
To determine if your study is an ACT requiring registration, use the checklist below. Additional considerations are elaborated on in the full ACT checklist at https://prsinfo.clinicaltrials.gov/ACT_Checklist.pdf, but these exceptions will be evaluated by the University of Iowa PRS administrators and IRB, who will inform the investigator when they apply. In addition to this checklist, many funding applications and FDA documents require specific language when a trial meets the definition of an ACT. More information on funding requirements can be found here, and these organizations may help in making the determination of an Applicable Clinical Trial. For a full document, please use the link at the bottom of this page.

More information can be found here.
3. Applicable Deadlines
|
Data Element |
Deadline for Updating (i.e., not later than the Specified Date) |
|---|---|
|
Study Start Date |
30 calendar days after the first subject is enrolled |
|
Overall Recruitment Status |
30 calendar days after a change in overall recruitment status
|
|
IRB Approval Status |
30 calendar days after a change in status |
|
Primary Completion Date |
30 calendar days after the clinical trial reaches its actual primary completion date |
|
Enrollment |
At the time the primary completion date is changed to "actual," the actual number of participants enrolled must be submitted |
|
Study Completion Date |
30 calendar days after the clinical trial reaches its actual study completion date |
|
Responsible Party, by Official Title |
30 calendar days after a change in the Responsible Party or the official title of the Responsible Party |
|
Responsible Party Contact Information |
30 calendar days after a change in the Responsible Party or the contact information for the Responsible Party |
|
Device Product not Approved or Cleared by US FDA |
15 calendar days after a change in approval or clearance status has occurred |
|
Record Verification Date |
Any time the Responsible Party reviews the complete set of submitted clinical trial information for accuracy and not less than every 12 months, even if no other updated information is submitted at that time. |
*Table Shared by Stanford University
4. NIH and ICMJE Reporting Requirements
NIH requires all clinical trials, regardless of study phase, to register on ClinicalTrials.gov. The NIH definition of a ‘Clinical Trial’ applies to all studies which are interventional and involve prospective assignment of subjects to a group or groups to test the effect of the intervention on a biomedical, health-related, or behavioral outcome. This definition differs from the federal regulations in that it is not required that a drug or device be involved. For example, a study of a behavioral intervention would meet the NIH definition of a clinical trial, but would not be an ACT under FDAAA 801 because it does not involve a drug or device. More information on NIH policies related to clinical trials can be found here.
Studies wishing to publish in International Committee of Medical Journal Editors (ICMJE) journals may be required to register their clinical trial. Investigators should be aware of the journals in which they would like to publish prior to starting a study, and familiarize themselves with the journal’s requirements.
| Element | Final Rule | NIH Policy | ICMJE Policy |
|---|---|---|---|
| Scope/Applicability |
Applicable clinical trials of FDA-regulated drug, biological, and device products and pediatric post-market surveillance studies of devices required by the FDA under the FD&C Act. Does not apply to phase 1 trials or small feasibility studies. Applicable clinical trials are (1) clinical trials of drug and biological products that are controlled, clinical investigations, other than phase 1 investigations of a product subject to FDA regulation; and (2) prospective clinical studies of health outcomes comparing an intervention with a device product against a control in humans (other than small feasibility studies) or any pediatric post-market surveillance studies required by FDA under the FD&C Act. Applies to public and private sector sponsors and other entities who meet the definition of a responsible party. |
All clinical trials funded wholly or partially by NIH. Includes phase 1 clinical trials and trials that do not involve any FDA regulated product such as trials involving only behavioral interventions. Applies to NIH-funded clinical trials where applications or proposals are received by NIH on or after the policy’s effective date. Applies to NIH-conducted clinical trials initiated on or after the policy’s effective date. |
All clinical trials which wish to publish in an ICMJE journal, or its affiliates, must register prior to enrolling the first subject. Currently, only registration is mandated. |
|
Timeframe for registration on ClinicalTrials.gov |
Not later than 21 days after enrollment of the first participant. |
Not later than 21 days after enrollment of the first participant. |
Prior to enrollment of first subject. |
|
Registration data elements to be submitted to ClinicalTrials.gov |
Elements defined in the final rule. Consists of descriptive information, recruitment information, location and contact information, and administrative data. |
Elements defined in the final rule. Consists of descriptive information, recruitment information, location and contact information, and administrative data. |
Elements defined in the final rule. Consists of descriptive information, recruitment information, location and contact information, and administrative data. |
|
Timeframe for results information submissions to ClinicalTrials.gov |
Not later than 12 months after primary completion date; possible delay of up to an additional 2 years for trials of unapproved products or of products for which initial FDA marketing approval or clearance is being sought, or approval or clearance of a new use is being sought. |
Not later than 12 months after primary completion date; possible delay of up to an additional 2 years for trials of unapproved products or of products for which initial FDA marketing approval or clearance is being sought, or approval or clearance of a new use is being sought. |
Not mandated in policy, but must meet the requirements of FDAAA 801. |
|
Results data elements to be submitted to ClinicalTrials.gov |
Elements defined in the final rule. Includes participant flow, demographic and baseline characteristics, outcomes and statistical analyses, adverse events, the protocol, and statistical analysis plan, and administrative information. |
Elements defined in the final rule. Includes participant flow, demographic and baseline characteristics, outcomes and statistical analyses, adverse events, the protocol, and statistical analysis plan, and administrative information. |
Elements defined in the final rule. Includes participant flow, demographic and baseline characteristics, outcomes and statistical analyses, adverse events, the protocol, and statistical analysis plan, and administrative information. |
|
Potential Consequences of Non-compliance |
|
|
|
*Table elements taken from https://www.nih.gov/news-events/summary-table-hhs-nih-initiatives-enhance-availability-clinical-trial-information
NIH and ICMJE affiliates (as well as many other journals) have even more stringent criteria for submitting to ClinicalTrials.gov. It is recommended that each PI be aware of each of these rules, or to practice following the most stringent of these to assure compliance.
5. Results Submission and Required Documents
All Applicable Clinical Trials are required to report results within 1 year of the date that the final participant was examined or received an intervention for the purposes of final collection of data for the primary outcome, whether the clinical study concluded according to the pre-specified protocol or was terminated. Additionally, all clinical trials utilizing NIH funds must report results information. In general, the law and federal funding entities require responsible parties to report summary results information for interventional studies of drugs, biological products, and devices within 1 year of the primary completion date of the study, regardless of the funding source, presence of statistically significant findings, or journal publication status. Prior to submitting results data, it is recommended you review the “How to Submit Your Results” guidance on ClinicalTrials.gov.
At the time the sponsor-investigator is ready to submit results, the Protocol and Statistical Analysis Plan (SAP) are required to be uploaded to the PRS record. If the SAP is included with the Protocol, then an additional document is not needed. These documents must be uploaded in PDF/A format, which can be converted Adobe Acrobat Pro or similar program. More information on this requirement can be found here.
6. Observational and Expanded Access Studies
Both Observational and Expanded Access Studies may be registered in ClinicalTrials.gov’s PRS; however, neither are considered Applicable Clinical Trials requiring registration. Please note that while expanded access studies are not Applicable Clinical Trials, if expanded access is offered for a drug or biologic, it must be noted in the record. Additionally, if an investigator both sponsors and manufactures an expanded access drug, they must create and maintain a record for that drug on ClinicalTrials.gov.
7. Protocol Registration and Results System (PRS)
The Protocol Registration and Results System (PRS) is the database where information published on ClinicalTrials.gov is entered. To access the PRS, investigators should go to register.clinicaltrials.gov. The PRS consists of two sections: the Protocol Section and the Results Section. The Protocol section is where registration information is entered and the results are entered in the results section along with information about participants and adverse events. Each section is broken down into modules where specific information is entered. The system, like HawkIRB, if very complex, but becomes easier to use after becoming acclimated with the modules. The PRS offers help and advice for completing each module via the ‘Definitions’ and ‘Help’ links on each page. These links work in much the same way as the HawkIRB help icons in that they describe exactly what the section is looking for. Additional information and training can be found on the ClinicalTrials.gov public site under the ‘Submit Studies’ tab. University of Iowa researchers should contact their PRS Administrator at ct-gov@uiowa.edu with additional questions. Please note the following University Policies when using the PRS:
- Users must input the IRB number (123456789) as the ‘Unique Protocol ID'
- Users should list themselves as ‘Sponsor-Investigator’
- The University of Iowa should not be listed as the sponsor for any study, unless otherwise instructed to do so
- The ‘Oversight’ module should list the IRB contact information as:
Board Status: Approved Approval Number: 123456789
Board Name: University of Iowa IRB 01
Board Affiliation: University of Iowa
Phone: 319-335-6564 Email: irb@uiowa.edu
Address:
Human Subjects Office / IRB
Hardin Library, Office 105
600 Newton Rd
Iowa City, IA 52242-1098
- Contact information should be for the study team and not the IRB
8. IRB Oversight
The University of Iowa IRB considers the ClinicalTrials.gov requirements in its approval. To help assure that investigators are in compliance with federal regulations, the IRB will notify investigators when these regulations apply. The University and PRS Administrator recommend that investigators submit the HawkIRB application first to avoid needing to make additional changes to the PRS record. In addition, the IRB reviewers and PRS administrator will assist in assuring the record is updated appropriately. The NCT number in the IRB application must be entered in VII.B.1.b of the IRB application prior to IRB approval to show that the ClinicalTrials.gov record has been created. Likewise, the ClinicalTrials.gov record must list the IRB number as the ‘Unique Protocol ID’ so that the records can be linked.
a. HawkIRB
Section VII.B of the HawkIRB application is where you will identify your clinical trial and find the link to the ClinicalTrials.gov database to register your study. VII.B.1 should identify ‘Clinical Trial’ as the study type, and additional questions can be answered, as appropriate. Changes to the HawkIRB system are currently underway to facilitate communication with the PRS system. Once the changes occur, much of the information will auto-populate into the PRS, making the maintenance and registration in the PRS easier for investigators.
b. VA Oversight of Investigator-initiated Clinical Trials
For studies where the VA is the primary study location or coordinating center of a study (listed in VII.A.10 of the IRB application), the study must be registered under the VA per their registration policy. Currently, the VA is requiring investigators who need to register at ClinicalTrials.gov under their institution to create and maintain their own institutional account in the Protocol Registration and Results System (PRS) for any records where the VA is the lead site. Accounts can be created by emailing a request to register@clinicaltrials.gov. Any study with both an IRB 01 and an IRB 03 application for the same study should look to VII.A.10 to identify which institution, the VA or UI, is the coordinating center of the study. Questions can be directed to Suzanne.kieffer@va.gov.
9. PRS Administrator
PRS administrators monitor records for the institution, provide help and feedback on using the system, create and monitor accounts for the PRS, and inform investigators of issues needing resolution in the PRS system. Additionally, the PRS administrator reviews study applications involving clinical trials for the IRB and communicates relevant information to them.
Individuals with questions or applying for an account with ClinicalTrials.gov through UI should contact:
Phone: 319-335-6564
Email: ct-gov@uiowa.edu
10. Resources
Getting Started – ClinicalTrials.gov Checklist
The ClinicalTrials.gov Checklist is an easy to use tool that allows investigators to prepare for registration and results reporting. The document easily lays out expectations and considerations, as well as provides links to useful materials such as planning templates.
ICMJE Data Sharing Statement
This guide will assist you in complying with the ICMJE Data Sharing policy.
The International Committee of Medical Journal Editors (ICMJE) requires:
- As of 1 July 2018 manuscripts submitted to ICMJE journals that report the results of clinical trials must contain a data sharing statement as described below.
- Clinical trials that begin enrolling participants on or after 1 January 2019 must include a data sharing plan in the trial's registration. The ICMJE's policy regarding trial registration is explained above. If the data sharing plan changes after registration this should be reflected in the statement submitted and published with the manuscript, and updated in the registry record.
ClinicalTrials.gov Redaction Guide
Understanding which information, if any, to redact is important when uploading required documents into ClinicalTrials.gov. This guide indicates when it is appropriate to redact, and when it should be avoided.
Redacting a document requires more than just crossing out words or placing a black box over the content to be removed. Documents must remove the text entirely so that it is no longer searchable, and this extends to metadata which may be stored in a document, hidden from sight. There are only a few programs that have been sufficiently vetted to redact information, but the University of Iowa recommends the use of Adobe Acrobat Pro.
ICTS Academy for Research Professionals Presentation Slides
This presentation is presented at the ICTS Academy for Research Professionals annually. It may be accessed at anytime as a resource for new PRS users, or as an education tool for existing users.
11. Office Hours
ClinicalTrials.gov Office Hours are held every Wednesday from 11:00 am – 12:00 pm in Hardin Library for the Health Sciences (HLHS) room 101.
The University of Iowa PRS Administrator is available to provide help and feedback to any ClinicalTrials.gov issues, such as:
- Registering a record
- Reporting results
- Responding to reviewer comments
- Identifying when registration is required or needed
- Training on using the Protocol Registration and Results System (PRS)
The IRB also offers office hours for IRB-specific issues. For more information on IRB Office Hours go here.
12. Helpful links
Grant information - https://grants.nih.gov/policy/clinical-trials/new-human-subject-clinical-trial-info-form.htm; https://grants.nih.gov/policy/clinical-trials.htm
Clinical Trials Policies - https://osp.od.nih.gov/clinical-research/clinical-trials/
Revised NIH definition of ‘Clinical Trial’ - https://grants.nih.gov/grants/guide/notice-files/NOT-OD-15-015.html
Final Rule information - https://prsinfo.clinicaltrials.gov/
NIH policy - https://grants.nih.gov/policy/clinical-trials/definition.htm
ICMJE policy - http://www.icmje.org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration.html

